Practice Problems in Pericyclic Reactions.
The following are a set of practice problems in Pericyclic Mechanisms, prepared by Henry Rzepa for the for a second year course in Pericyclic Reactions at the Department of Chemistry, Imperial College. Each of these problems is intended to take between 1-3 hours to solve. They are NOT typical of examination questions! Please note also that the compound numbers are not consecutive between questions.
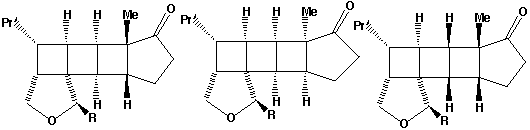
Qu 31
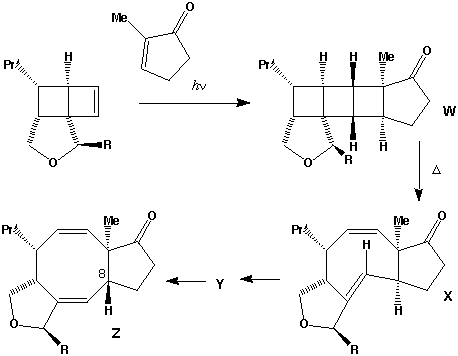
- The π2s + π2s cycloaddition is a 4n electron photochemical allowed process proceeding with suprafacial addition across both alkene components (M. L. Randall, P. C. K. Lo, P. J. Bonitatebus and M. L. Snapper, J. Am. Chem. Soc., 1999, 121, 4534).
- The alternate regioisomer
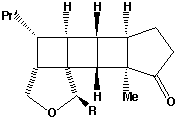 of W is not formed when R=Me (or larger) due to steric hindrance between the two methyl groups (see molecular models).
of W is not formed when R=Me (or larger) due to steric hindrance between the two methyl groups (see molecular models).
- Three other so-called endo/exo stereoisomers of W are all consistent with the π2s + π2s cycloaddition. Molecular modelling can be used to establish the relative energies of all four isomers. The one actually formed is found to be lower in free energy than the other three, largely a result of minimisation of the steric repulsions between the R group and other atoms. Other stereoisomers could result from π2s + π2a addition across the two alkenes to give a trans ring geometry, but such stereochemistry would not be consistent with the rules for a 4n electron photochemically mediated cycloaddition.
-
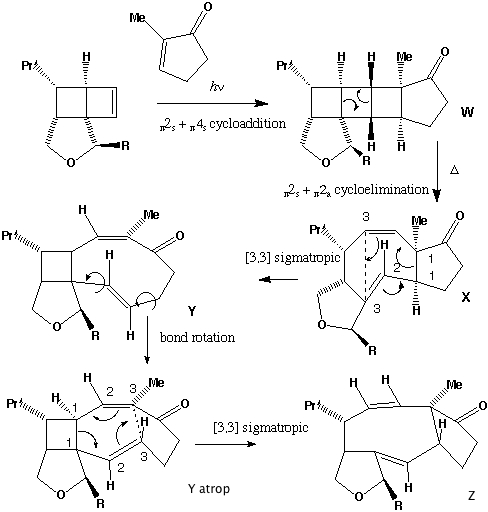
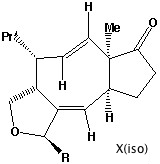
- Thermolysis requires a π2s + π2a retro- cycloaddition (4n electrons, Mobius transition state with an antarafacial component). The antarafacial component can be located on either of the forming two alkene bonds. For isomer X, this produces the cis alkene shown, twisting the original trans relationship of the two hydrogens on these two carbons by 180 degrees.
-
- Locating the antarafacial component on the other possible forming alkene results in the following isomer of X:
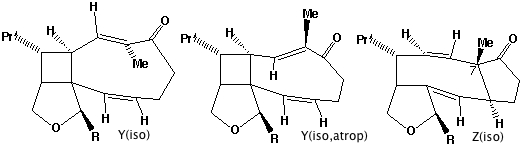
- Compound X has the characteristic 3-single/two-double bond pattern associated with the [3,3] sigmatropic Cope rearrangement. This is actually possible in two directions, but we concentrate on the one that creates an sp2 centre at C-8, the atom at which inversion of configuration is observed to occur. This produces a flexible 9-membered ring, which can flip the trans alkene bond by atropisomerism (rotation) about the adjacent single bonds, to produce a conformer with the C-8 hydrogen pointing up. Reverting the [3,3] sigmatropic reaction using this conformation will now reform the 5-8-5 ring system but with the correct stereochemistry at C-8.
- Repeating the sequence of events on the isomer of X produces the following compounds, i.e. bond rotation about the trans alkene and [3,3] Cope reclosure now inverts the configuration at the C-7 carbon. Molecular modelling indicates this pathway is only about 20 kJ/mol higher than the observed one, suggesting this might be a minor (unobserved) product of this reaction.
Qu 32
- 4n+2, Huckel 2+4 cycloaddition to give [A], followed by 4n+2 Huckel cycloelimination. Two possible regio-isomers are possible for [A] (bonus mark if you spotted this).
- Electrocyclic 4n Mobius ring opening (as per lectures) followed by rotation around the single bond indicated and electrocyclic ring closure to distribute the deuterium atoms as shown.
- Two successive electrocyclic reactions, the first 4n via Mobius to give the trans dimethyl stereochemistry, the second 4n+2 via Huckel to give the cis-ring fused system. Then 4n+2 Huckel cycloaddition (two possible stereoisomers, but one is more sterically hindered than the other) followed by cycloelimination of a cyclobutene, which then opens in a 4n Mobius electrocyclic reaction to give the trans hexadiene. This problem becomes more tractable if the starting material is redrawn into a cyclisable form by rotating about formally single bonds, hence the hint!
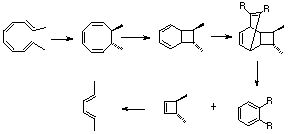
- A Mobius 4n 2+2 cycloaddition typical of that found for many ketenes gives a cyclobutene, which opens via an electrocyclic 4n Mobius to a diene. A second 4n+2 electrocyclisation is followed by a [1,7] sigmatropic shift of the H atom via a Mobius (antarafacial) migration. If traces of acid or base are present, then a normal catalysed keto-enol tautomerism will occur instead.
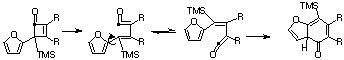
Qu 33
Photolysis of U results in a carbene, which undergoes a 4n [1,2] sigmatropic shift (the Wolff rearrangement) to give the ketene V. This undergoes a p2s + p2a cycloaddition (Mobius transition state) with the alkyne to give the cyclobutenone W, which in turn opens by an electrocyclic 4n reaction (Mobius transition state) to give either X (conrotation in one direction) or XÕ (conrotation in the other direction). Only X is capable of further 4n+2 electrocyclisation (Huckel transition state) to give Y, which then tautomerises to give the phenol Z. For more details, see R. L. Danheiser et al, J. Am. Chem. Soc., 1990, 112, 3093. 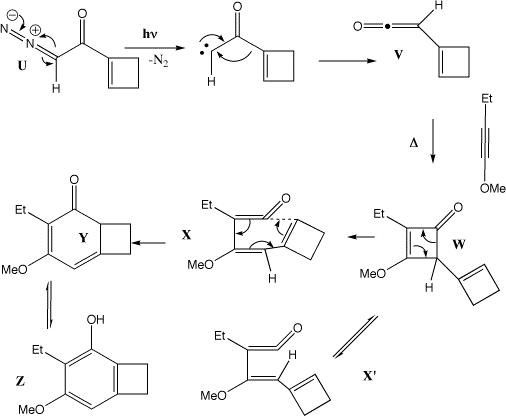
Qu 34
A to B is a typical [3,3] sigmatropic rearrangement (two double bonds separated by three s bonds) and is a 4n+2 thermal process proceeding via a Huckel transition state with suprafacial stereochemistry. The enol so formed rapidly tautomerises to the ketone C, which then undergoes an ene reaction (two s bonds formed, one broken) to form D, also a 4n+2 Huckel reaction. Either one of the two hydrogen atoms on the prochiral CH2 group (differentiated as Pro-R and Pro-S) can be transferred resulting in E or Z double bond isomers, via a transition state the more stable of which leads to D and the less stable to E. D/E again have the pattern of two double bonds separated by three sigma bonds, which allows another [3,3] sigmatropic reaction to occur, via a chair 4n+2 Huckel transition state in which the new C-C bond is formed suprafacially to create two new chiral centres in F/H (see models). This rapidly and non-stereospecifically cyclises to the final product G. The other double bond isomer E which [3,3] cyclises via another chair transition state to H does so with the opposite configuration at the benzylic carbon (see the 3D models to really convince yourself this is true). For more details, see L. Barriault and I. Denissova, Organic. Letters, 2002, 4(8), 1371-1374. 3D models for these transition states can be viewed at http://www.ch.ic.ac.uk/local/organic/tutorial/rzepa6/. 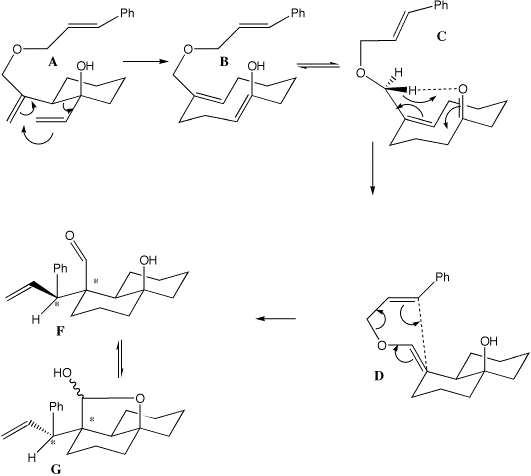
Qu 35
See here.
Qu 36
See here.
Qu 37
See here.
THE END
© Henry S. Rzepa, 1978-2014. Hide|show Toolbar.