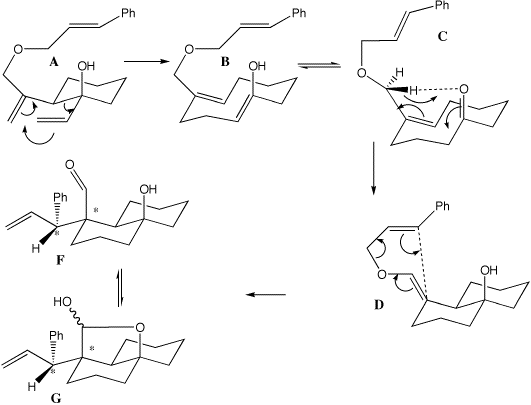
Following a conventional [3,3] Cope rearrangement of A to B, with subsequent tautomerisation to C, a more unusual Ene reaction to form D is observed, a component of which is the trans annular ring closure. Abstraction by the carbonyl oxgyen can be of either of the two O-CH2 hydrogen atoms (Pro-R or Pro-S ) resulting in two different transition states and of course two possible products differing in the E/Z configuration of the resulting double bond . Quantitative molecular models (AM1) predict 2 kcal/mol difference between these two transition states, in accord with the observed stereospecificity.
| TS leading to D (-11.2 kcal/mol) | TS leading to E (-9.2 kcal/mol) | ||
|---|---|---|---|
| in out Display spacefill ball & stick stick wireframe |
in out Display spacefill ball & stick stick wireframe |
||
The reaction completes with a [3,3] Claisen rearrangement, in which new chiral centres at the carbon atoms defining the termini of the forming C-C bond are formed stereospecifically. Quantitative models show why this should be. The [3,3] rearrangement proceeds via a Chair transition state ( ) with the two new chiral centres highlighted in yellow(). In this model, the new C-C bond is forming on the face of the C=C system () antiperiplanar to the C-OH group. The alternative isomer, in which the new C-C bond is formed from the synplanar face of the alkene, is much more sterically hindered and the transition state has a much higher energy (-15.8 kcal/mol).
| TS leading to F (-26.6 kcal/mol) | TS leading to H (-23.8 kcal/mol) | Synplanar TS (-15.8 kcal/mol) | |||
|---|---|---|---|---|---|
| in out Display spacefill ball & stick stick wireframe |
in out Display spacefill ball & stick stick wireframe |
in out Display spacefill ball & stick stick wireframe |
|||