Theoretical Explanation.
 Our story starts with Évariste Galois (1811-1832) who founded the theory based on the symmetries of permutations which we now know as group theory. The original explanation of Woodward and Hoffmann based on the nature of the highest occupied molecular orbitals was formalised with the help of Galois' group theory by Longuet-Higgins and Abrahamson (DOI: 10.1021/ja01087a033, see also 10.1021/ja01087a033). This involved generating a so called orbital correlation diagram for the reaction under consideration, and then carrying out the reaction in such a manner that the group theoretical symmetries of the reactant and product orbitals matched exactly. Such an approach, whilst theoretically more rigorous, is not readily applicable to the majority of more complex reactions which have no formal symmetry. Two much simpler methods have been subsequently outlined which avoid this problem. The first these, based on transition state aromaticity, will be expanded in more detail, whilst the second (based on frontier orbitals) will only be described briefly in the next lecture.
Our story starts with Évariste Galois (1811-1832) who founded the theory based on the symmetries of permutations which we now know as group theory. The original explanation of Woodward and Hoffmann based on the nature of the highest occupied molecular orbitals was formalised with the help of Galois' group theory by Longuet-Higgins and Abrahamson (DOI: 10.1021/ja01087a033, see also 10.1021/ja01087a033). This involved generating a so called orbital correlation diagram for the reaction under consideration, and then carrying out the reaction in such a manner that the group theoretical symmetries of the reactant and product orbitals matched exactly. Such an approach, whilst theoretically more rigorous, is not readily applicable to the majority of more complex reactions which have no formal symmetry. Two much simpler methods have been subsequently outlined which avoid this problem. The first these, based on transition state aromaticity, will be expanded in more detail, whilst the second (based on frontier orbitals) will only be described briefly in the next lecture.
1. Conservation of Orbital Symmetry (Longuet-Higgins and Abrahamson)
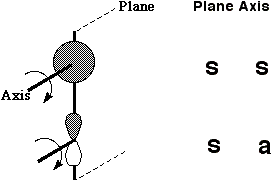
Let us first define the symmetry properties of a 1s and a 2p orbital with respect to a plane of symmetry or an axis of symmetry as shown below;
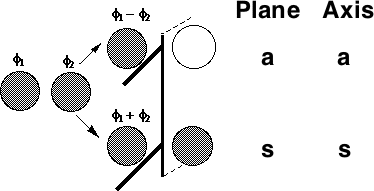
One can take this one step further by considering the symmetry properties of molecular orbitals formed by the overlap of two or more atomic orbitals;
MOs formed from two overlapping σ orbitals:
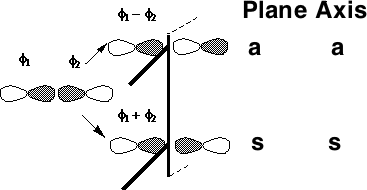
MOs formed from two overlapping p-orbitals (σ bonds):
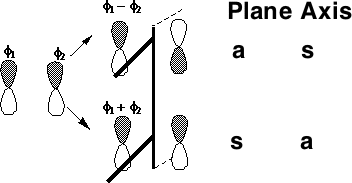
MOs formed from two parallel overlapping p-orbitals (π bonds):
We can now use these basic orbitals to construct the relevant molecular orbitals for two interconverting molecules, cyclobutene and butadiene, with the purpose of following how these two sets of orbitals change when one molecule is converted into the other. Note particularly that we need only construct the MOs explicitly involved in the reaction; most of the σ framework remains unchanged and no orbitals derived from this need to be considered:
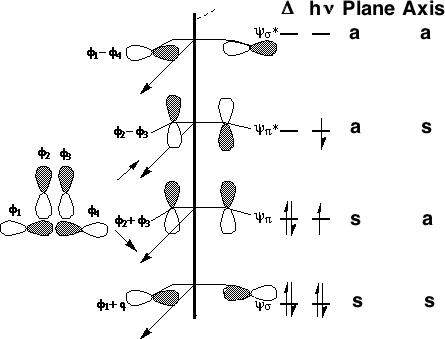
(click on the diagrams above or below to see the actual molecular orbitals)
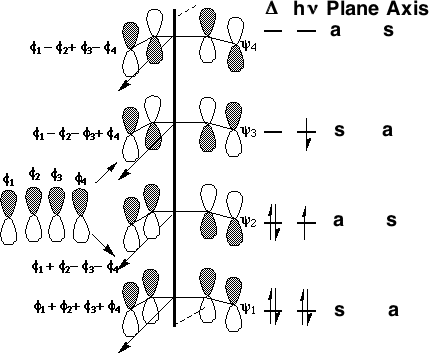
In order to interconvert cyclobutene and butadiene, the four MOs labelled ψ1, ψ2, ψ3, ψ4 must be converted into ψσ, ψπ, ψπ*, ψσ*. There are two stereochemically distinct ways in which this might be accomplished;
Conrotation:
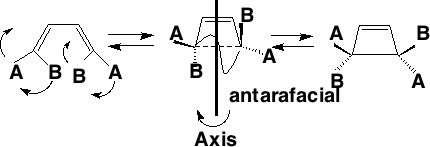
Disrotation:
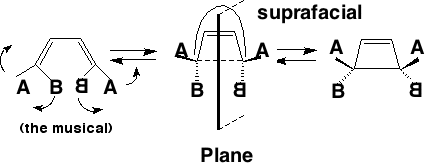
This enables a correlation diagram for the reaction to be constructed, according to the following rules: no two orbitals of the same symmetry can cross during the reaction, whilst orbitals of different symmetry can cross. The favoured pathway is the one which results in a product of the same electronic excitation as the reactant (green).
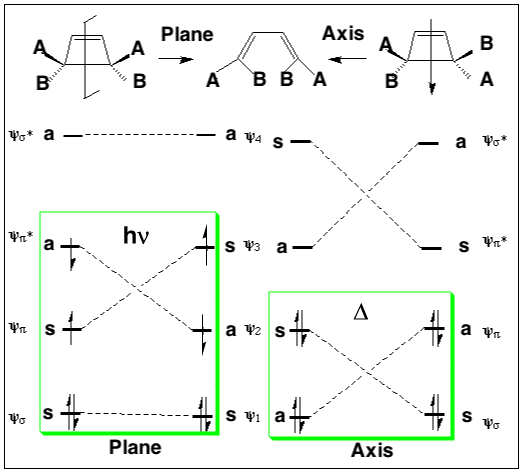
Pathways which result in the product being formed in a higher electronic state than the reactant are said to be "forbidden" (red).
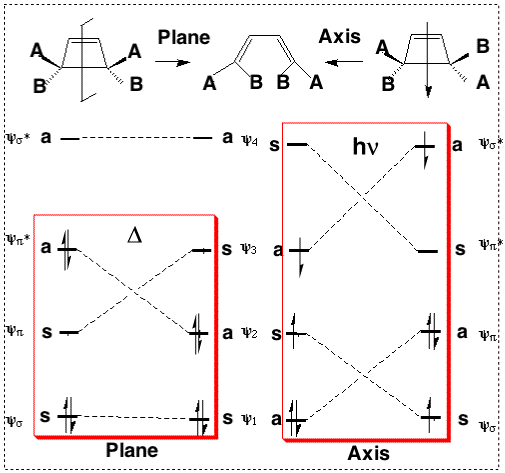
Whilst this rule is normally followed fairly well for ground states, it can be overturned when for example steric or geometrical strain in the "allowed" pathway promotes the "forbidden" route. The situation is actually more complex for photochemical reactions, and much recent evidence suggests that the Woodward-Hoffmann rules are not always followed. These correlation diagrams can be generalised for any electrocyclic reaction with appropriate symmetry. However, correlation diagrams are less readily applied for reactions with no symmetry and a method more tolerant of broken symmetry is required. In the next section we will introduce such a method.
© Henry S. Rzepa, 1978-2014. Hide|show Toolbar.