The most common example is the Diels Alder cycloaddition (for a review, see here). Two π systems are involved, one contributing 4π electrons, the other 2π electrons. The total count is 6 (4n+2, n=1) and since the reaction is thermal, it must proceed via Hückel topology involving only suprafacial components (or, hypothetically, two antarafacial components, but this is geometrically impossible here).

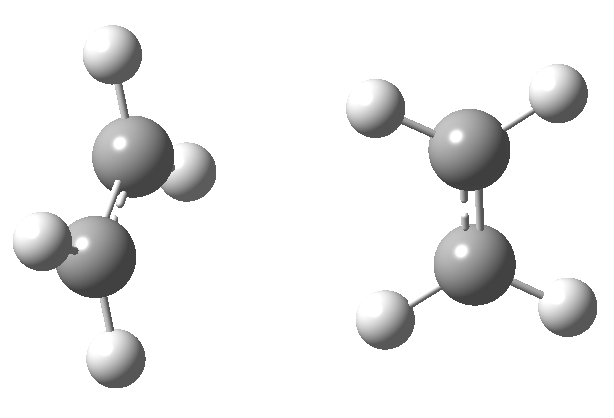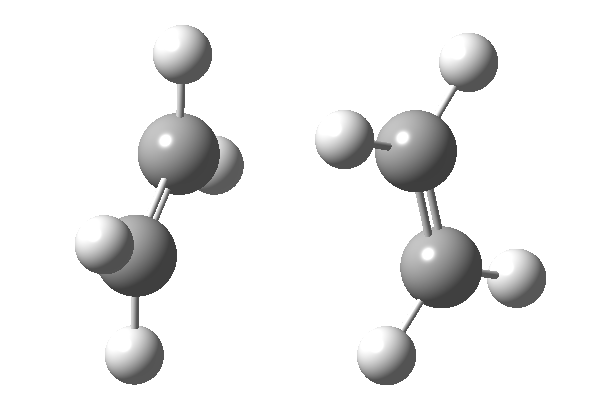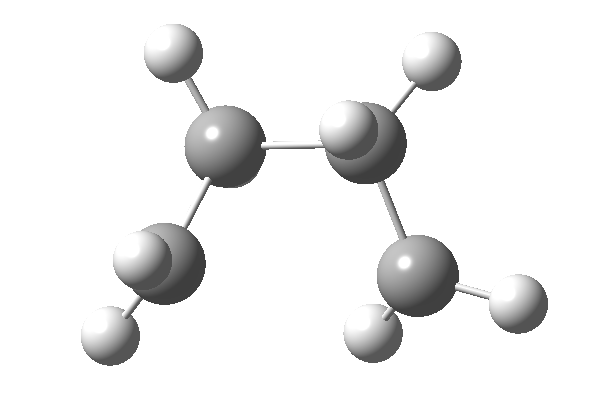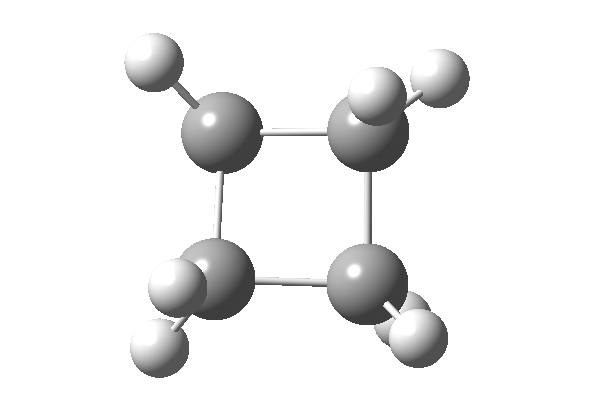
![]() These are considered as follows. Each π system forms two new σ bonds. These two new bonds can either form on the SAME face of one π system (suprafacial) or from opposite faces of one π system (antarafacial). A system of nomenclature has been developed to describe this. Hence the system below is classified as a π4s + π2s cycloaddition (i.e. both bonds forming on the bottom face of the diene = π4s and both bonds forming on the top face of the alkene = π2s, the so-called exo form). An isomer in which the bonds form to the bottom face of the alkene is also π2s (the endo isomer) is actually predicted to be slightly more stable (as a transition state) than the exo form. For a brief discussion of the application of frontier orbital method to cycloaddition reactions, see here.
These are considered as follows. Each π system forms two new σ bonds. These two new bonds can either form on the SAME face of one π system (suprafacial) or from opposite faces of one π system (antarafacial). A system of nomenclature has been developed to describe this. Hence the system below is classified as a π4s + π2s cycloaddition (i.e. both bonds forming on the bottom face of the diene = π4s and both bonds forming on the top face of the alkene = π2s, the so-called exo form). An isomer in which the bonds form to the bottom face of the alkene is also π2s (the endo isomer) is actually predicted to be slightly more stable (as a transition state) than the exo form. For a brief discussion of the application of frontier orbital method to cycloaddition reactions, see here.
| endo | exo |
|---|---|
 |
 |
An example of an enzymically catalysed Diels-Alder reaction is the unique enzyme extracted from Alternaria Solani which catalyses a π4s + π2s cycloaddition in prosolanapyrone (III) with very high exo-selectivity (the normal uncatalyzed selectivity is endo). The structure of this enzyme was reported here.


This highly active (indeed controversial, see here) area includes discussion of Diels-Alderases, Theozymes, antibodies and Biosynthetic Diels Alder.
Diels Alder additions have also been observed catalysed within the cavities of certain organic crystal structures. In this example (10.1021/ja964198s) one molecule of cyclohexadiene is encapsulated inside a cavity formed from four ligand molecules, along with TWO molecules of ethyl methacrylate either side, one of which adds in Diels Alder fashion. The specificity for endo is enhanced by the crystal cavity.![]()

Such reactions can also be catalysed using water as a solvent (10.1021/ja0010249), the catalysis arising in part due to specific hydrogen bonds which are strongest in the transition state.
![]() This next example is a 4 electron (4n) cycloaddition reaction,
This next example is a 4 electron (4n) cycloaddition reaction,
 which proceeds photochemically (DOI: 10.1021/ja01030a066) via a Hückel topology (π2s + π2s). The ratio of all-cis (endo) to the exo form was unexpectedly found to be 1.3:1, indicating attraction rather than repulsion between the methyl groups. Photochemical reactions are nowadays recognised as being controlled by the properties of a conical intersection (the lowest energy point at which the ground and excited state "touch") rather than a transition state, although the selection rules derived earlier are in fact often adhered to.
which proceeds photochemically (DOI: 10.1021/ja01030a066) via a Hückel topology (π2s + π2s). The ratio of all-cis (endo) to the exo form was unexpectedly found to be 1.3:1, indicating attraction rather than repulsion between the methyl groups. Photochemical reactions are nowadays recognised as being controlled by the properties of a conical intersection (the lowest energy point at which the ground and excited state "touch") rather than a transition state, although the selection rules derived earlier are in fact often adhered to. 
Thermally, the dimerisation of e.g. ethene is predicted by the selection rules to proceed as a π2s + π2a, the subscript a indicating an antarafacial component must be present on one alkene. However, there is a problem; an antarafacial mode implies a Möbius like twisting of 180° in the π system of the ethene, but ethene has only two carbons and cannot be twisted so much! A quantitative model indicates that only about 130° of twisting can actually be induced, of which about 80° occurs on one ethene and 50° occurs by twisting one ethene with respect to the other. Strain is also relieved by forming the two bonds at different rates.
The trans alkene shown below has sufficient driving force to actually indulge in this rather twisted transition state. Note that only the two strained trans double bonds react; the more stable cis double bonds do not participate in the reaction, although they do help stabilize the transition state. See here for the original report, and here for an update which implicates some contribution from biradicals in the process, as do high level calculations.


The antarafacial component is present only in the transition state, and it has the effect of giving the more sterically hindered product (see DOI: b3sxdz). It was noted above that one way of relieving strain induced by an antarafacial component is to make the two forming bonds of unequal length. In this example, this inequality allows the two large groups (t-butyl and phenyl) to avoid each other in the transition state, even though this is not possible in the final product. At the transition state, this isomer shown here is 3.0 kcal/mol lower in free energy than the unformed C-Ph epimer.
In extreme of bond asymmetry, which the ketene above is tending towards, this corresponds to forming a carbocation and carbanion. This of course is now a stepwise rather than concerted reaction and is no longer a pericyclic process. Many 2+2 cycloadditions are thought to proceed in this manner, especially in more polar solvents which stabilise the intermediate zwitterions (and which non-polar solvents suppress). In other systems, such asymmetry corresponds to the formation of so called "biradical intermediates", and recent evidence suggest these routes may be more common than hitherto suspected!




Ten electrons (4n+2, n=2) can also participate in thermal cycloadditions (DOI: 10.1021/ja00853a063) such as the reaction between tropone (six electrons) and cyclopentadiene (four electrons). The reaction is suprafacial on both the 6 and the 4 components (π4s+π6s):


However, we can again see an incursion of unequal bond lengths at the transition state, and this might be a feature of all cycloadditions with 10 or more electrons.
The largest number of electrons in a thermal cycloaddition has been proposed as 16, in a π2s + π14a reaction (See DOI: 10.1016/0040-4039(81)80058-5). Since 16 electrons = 4n (n=4) this requires one antarafacial component.

One point to note is that the antarafacial component could in principal occur on either alkene, but in practice the geometrical distortion is such that either the more strained or the larger component bears the antarafacial mode. This system is also likely to feature unequal lengths of the forming bonds.
 This system was then subjected to X-ray analysis to determine the structure of the cyclobutadiene. The controversy is over whether the second photolysis did take place at all, or if it did, whether a thermal π2s - π4s cycloaddition reaction occurs rapidly enough to destroy the cyclobutadiene and reform compound 2.
This system was then subjected to X-ray analysis to determine the structure of the cyclobutadiene. The controversy is over whether the second photolysis did take place at all, or if it did, whether a thermal π2s - π4s cycloaddition reaction occurs rapidly enough to destroy the cyclobutadiene and reform compound 2. 
A subsequent intramolecular π2s + π4s Diels Alder cycloaddition gives the final product (note how in this transition state, one of the forming C-C bonds forms at a significantly faster rate than the other, it being "tethered" by the adjacent five membered ring, whilst the other end is more floppy).

The bonds formed in this overall sequence can be inspected by clicking on the diagram below:
Eight stereogenic centres are formed from the starting achiral polyene, implying that 128 stereoisomers (+ their 128 enantiomers) could be formed. In fact, the pericyclic selection rules reduce this number (not completely down to one of 128, since the selection rules do not specify whether the Diels-Alder step is endo or exo). The only isolated form has the relative stereo-chemistries shown in the model above. But in the absence of any additional chiral auxiliary, this stereoisomer is formed as a racemic mixture of it, and its enantiomer.
