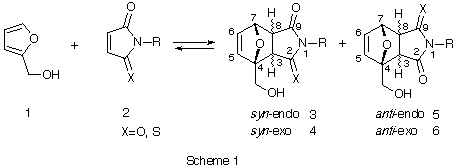
Furfuryl alcohol underwent Diels-Alder reaction with N-substituted thionomaleimides to afford high yields of cycloadducts with syn predominance, which can be attributed to the intermolecular hydrogen bonding between -OH -- S=C at the transition state.
Introduction
The Diels-Alder reaction is widely used to construct six-membered carbon skeletons.[1] Reaction of an unsymmetrical diene with unsymmetrical dienophile can give rise to two regioisomeric adducts. Many attempts have been made to control stereo- and regio-selectivity of the reaction, and are successful mainly by use of Lewis acid catalyst or introduction of chiral substituent to the synthons.[2] On the other hand, drug-receptor interaction in vivo depends on the ability of molecules to bind and discriminate between each other. This phenomenon is explained in terms of weak and noncovalent interactions such as hydrogen bonding, hydrophobic interaction, van del Waals interaction, dipole-dipole interaction, charge transfer complexation and so on.[3] In this connection, we found a periselective 1,3-dipolar cycloaddition of pyridine N-oxides with phenyl isocyanates, which proceeds via the charge-transfer complexation.[4] In our preliminary experiments on the cycloaddition of furfryl alcohol with N-phenylthionomaleimide in refluxing benzene, syn-cycloaddition predominantly occurred (syn : anti = 4 : 1), which might be attributable to the hydrogen bonding. As far as we know, there is little research showing control of the regioselectivity of pericyclic reactions using such weak interactions.[5] We now report an attempt to control the selectivity of Diels-Alder reaction by intermolecular OH -- S=C hydrogen bonding.
Product research
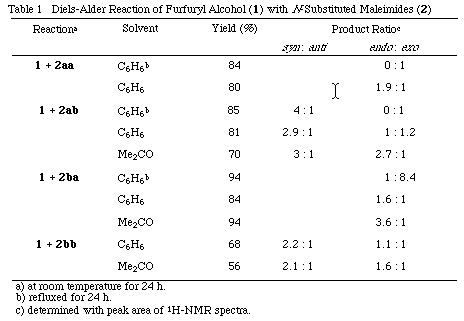
Furfuryl alcohol (1) was allowed to react with N-phenylmaleimide (2aa), N-phenylthionomaleimide (2ab), N-methylmaleimide (2ba) and N-methylthionomaleimide (2bb), respectively under the conditions given in Table 1.
As shown in Table 1, the reaction of 1 with 2 proceeded smoothly at room temperature to give corresponding adducts (3-6) in high yields. It is noteworthy that the reactions of 1 with thionomaleimides (2ab, 2bb) predominantly gave syn-adducts (3 and 4). The formation ratio ranged between 4.2 to 2. To investigate such syn predominance in Diels-Alder reaction, 9-(hydroxymethyl)anthracene (7) was allowed to react with 2ab in C6H6 at room temp. for 6 d to give a 2.4:1 mixture of syn and anti adducts (syn-8 and anti-8) in 90% yield.
In this reaction, the endo selectivity rises as the solvent polarity increases. In refluxing benzene, the formation ratio of exo adduct increased, which can be attributed to the thermodynamic stability of the adducts.
Kinetic study
This reaction involves equilibrium between starting materials and products. We performed kinetic study using large excess (100 equiv.) of 1, by which we can treat the reaction as a pseudo-first-order reversible reaction.[6]
No significant increase of reaction rate with increase of solvent polarity can be observed for either the forward or the reverse reaction, suggesting that the reaction of 1 with 2ab proceeds in a concerted manner.
The reaction of 1 with 2ab showed a remarkably negative activation entropy, suggesting that the reaction is entropically controlled. In this situation, the reaction proceeds via a very restricted transition state.
MO consideration for selectivities
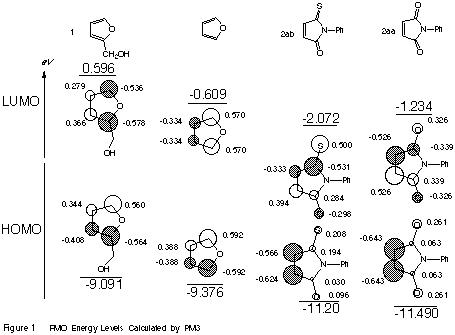
PM3[7] calculation of 1 and 2 were performed to investigate the reaction from frontier molecular orbital (FMO) point of view by the MOPAC[7] (version 6.02) molecular orbital package which was locally modified for Sun FORTRAN 1.2 on a Fujitsu S-4/2 engineering workstation.
As can be seen in Figure 1, the dominant orbital interaction operates between the HOMO of 1 and the LUMO of 2ab. The forward reaction of 1 with 2aa at 36 oC in AcOEt proceeds 2.4 times more quickly than that of 1 with 2ab. Taking the energy gaps between the FMOs of the synthons (1, 2ab and 2aa) into consideration, 1 is expected to react more smoothly with 2ab than with 2aa. However, the kinetic result mentioned above is contrary to expectation. The coefficients at the reaction site (C=C) of 2ab are smaller than those of 2aa, which may explain the experimental result.
Next, mention should be made of regioselectivity. Inspection of the magnitude of the FMO coefficients at the reaction site of 2ab and 1 predicts an anti cycloaddition, which is in disagreement with the experimental result, indicating that simple FMO consideration canot fully explain the syn predominance of the reaction of 2 toward 1. It is worth noting that the lone-pair orbital on the sulfur atom has a large coefficient in the plane of maleimide ring of 2ab, suggesting that the observed syn predominance might be explicable by the hydrogen bonding between CH2OH of 1 and C=S of 2. To obtain further evidence, we performed MO calculation with COSMO[8] (Conductor-like Screening Model) keyword in MOPAC93[9] to approximate the effect of a solvent model surrounding the molecule. The COSMO calculation for maleimide revealed that significant polarization occurs in water compared with in the gas phase, namely, a markedly greater charge on the sulfur atom and a significant increase in the dipole moment. This result favours hydrogen bond formation between OH and S=C moieties. The visible absorption spectrum of 2 in MeOH showed only a very small blue shift, indicating that the hydrogen bonding between solvent and 2 is not so strong.
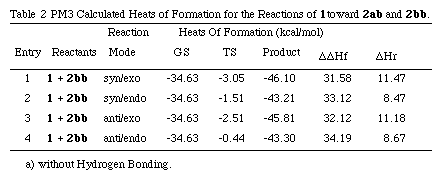
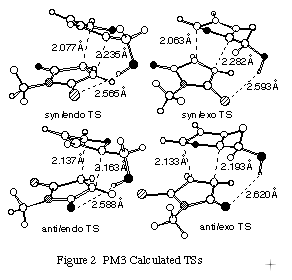
Next, to obtain a quantitative clue for the reaction mechanism for the reaction of 1 with 2, we performed PM3 simulation for the cycloaddition. The PM3 transition states (TS) for the cycloaddition of 1 with 2 were located using the TS method or SADDLE routine[10] followed by the NLLSQ method[11] implemented in MOPAC and characterized by establishing that the Hessian (force constant) matrix had only one negative eigenvalue.[12]
TS calculations for the possible cycloaddition modes of 1 toward 2bb and 2ba were performed. In the reaction of 1 with 2bb, the syn-TSs are more stable than the anti- ones, which agree to the result of product research.
As mentioned above, the forward reaction of 1 with 2aa at 36 oC in AcOEt proceeds 2.4 times more quickly than that of 1 with 2ab. The TS calculations could reproduce the kinetic results very well.
Among the adducts, the exo adducts are more stable in heat of formation than the exo ones, which is consistent with the synthetic result in refluxing benzene. [[Query exo/exo???]]
It was reported that the polarity of the solvent determine the ratio of endo/exo products in kinetically controlled Diels-Alder reactions.[13] The more polar solvents favour endo addition. It is well known as the omega value.[13] In this study, the endo selectivity is inclined to increase as the solvent polarity increases (Table 1). In this connection, we performed TS calculations with COSMO keyword to test whether PM3 method can reproduce such a solvent effect or not. The exo TSs in both CHCl3 and MeOH are more stable than that of the endo ones.
Conclusion
Intermolecular hydrogen bonding between -OH -- S=C controls the selectivities of cycloaddition between 1 and 2ab or 2bb. TS calculations reproduced the syn/anti selectivity very well. However, the endo/exo selectivity could not be reproduced by PM3 TS calculations.
Acknowledgement
We thank the members of the Analytical Center of Kumamoto University for microanalyses and spectral measurements.