A formal synthesis of perhydrohistrionicotoxin
Department of Chemistry, North Carolina
State University, Raleigh, NC 27695-8204, USA
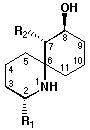
In 1971, Wiktop and coworkers reported the isolation of (-)-histrionicotoxin
(1), (Figure 1), an alkaloid with a skeleton of a
piperidine-containing spirocyclohexane unit, from skin extracts of the "arrow
poison frog" Dendrobates histrionicus.1 Both 1 and
its perhydroderivative 2 are equally bioactive alkaloids, which are
useful tools in neuroscience for the study of the mechanisms involved in the
transynaptic transmission of neuromuscular impulses.2 In recent
years, the synthesis of the histrionicotoxin family has attracted attention not
only due to its interesting biological activity in conjunction with its
scarcity (ca. 200 ug per frog), but also due to the challenge of
making this unique azaspirocyclic skeleton with a certain spatial arrangement
of amino and hydroxy groups.
1 R1 = CH2-HC = CH-C...triple...CH
R2 = HC = CH-C...triple...CH
2 R1 = C5H11
R2 = Bu
Figure 1 Structures of histrionicotoxin (1) and
perhydrohistrionicotoxin (2).
Since the first total synthesis of (+/-)-perhydrohistrionicotoxin (PHTX) 2
by Corey in 1975,3a organic chemists have continued to
investigate different approaches toward the stereoselective construction of
this azaspiro[5.5]undecane skeleton. Numerous reports have appeared on
synthetic studies in this series;3,4 however, only four of them have
addressed the problem of asymmetric synthesis of the alkaloids.4
The successful asymmetric reactions involving 2,3-dihydro-4-pyridones
developed by the Comins group5 have been applied to the synthesis of
optically pure alkaloids. By careful consideration, a synthetic plan was
designed to prepare (-)-PHTX (2) utilizing a photocycloaddition of a
chiral 2,3-dihydro-4-pyridone as a key step. The azaspiro skeleton of 2
would be built in a stereoselective manner via a short and convergent
route. To test this approach a racemic synthesis was initiated according to
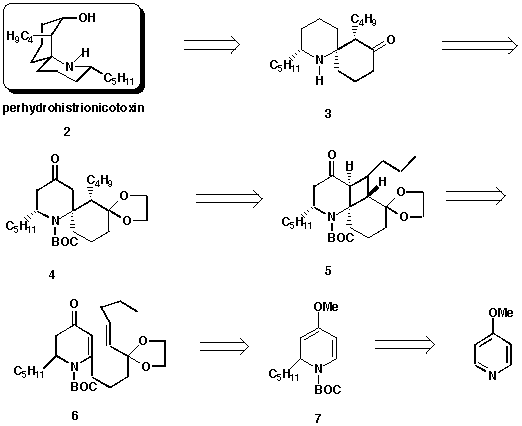
the plan outlined in Scheme 1.
Scheme 1
The natural product (+/-)-(PHTX) (2) would be synthesized by the
reduction of amino ketone 3 with LiAl (O-But)3H in
THF.4a Amino ketone 3 would be generated by deprotection of
the ketal and tert-BOC groups of 4. Compound 4 would be the
product of a ring-opening reaction of the photoadduct 5 using the
reducing agent SmI2. Intermediate 5 would arise by the intramolecular
photocycloaddition of a 2,3-dihydro-4-pyridone with a nine-carbon tether side
chain containing a C=C bond and a protected ketone functionality at the
position next to the double bond. The photosubstrate 6 would be
prepared by alkylation of 4-methoxy-2-pentyl-1,2-dihydropyridine 7 at
the C-6 position with an alkyl iodide, containing the whole side chain, via
ortho-lithiation and subsequent electrophilic substitution.6 The
introduction of a pentyl group at the C-2 position of 7 would be
obtained by pentyl Grignard addition to an N-acylpyridinium salt.
According to the synthetic strategy for PHTX (2) (Scheme 1), a linear
nine-carbon side chain at C-6 of 6 was required. We prepared the iodide
8 using classical chemistry, and began studies directed at joining this
unit with dihydropyridine 7 to give dihydropyridone 6 on acidic
workup.
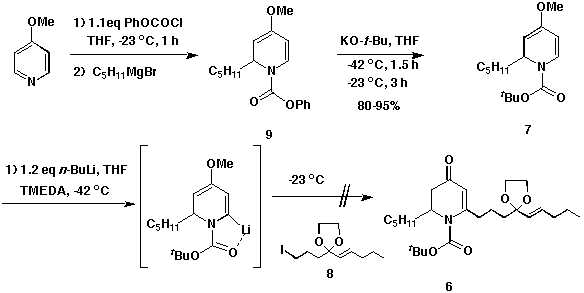
Readily available 4-methoxypyridine was treated with phenyl chloroformate and
subsequently pentyl Grignard. The product 9 was treated with
potassium tert-butoxide to make the N-BOC derivative of
1,2-dihydropyridine 7. The a-lithiation of 7 followed by
treatment with the electrophile 8 resulted in recovery of unreacted
starting material. Although this reaction was repeated with prolonged reaction
times and various other conditions, we were not successful in generating the
desired product 6.
Scheme 2
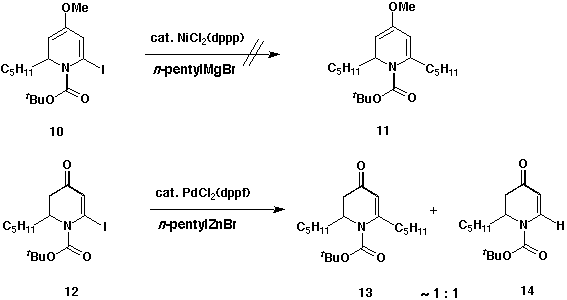
Several other methods to introduce the required side chain at C-6 of the
pyridone 6 were attempted. One approach involved a coupling reaction of
6-iodo-1,2-dihydropyridine 10, prepared by iodination of dihydropyridine
7, with excess alkyl Grignard reagent catalysed by
NiCl2(dppp).7 Unfortunately, this reaction offered less than 5% of
cross-coupled product 11. Another was a coupling reaction between
6-iodo-2,3-dihydro-4-pyridone 12, prepared from the hydrolysis of 10,
and an alkylzinc reagent utilizing the palladium ctalyst,
PdCl2(dppf).8 The material isolated from this reaction was a mixture
of both 13 and reduced compound 14 in the ratio of approximately
1:1. Since attempts at introducing the entire side chain 8 were
unsuccessful, the strategy for incorporation of the tethered olefin was
changed.
Scheme 3
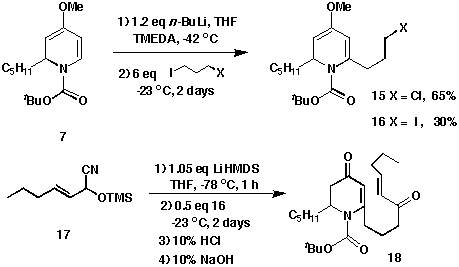
It was found that alkylation of dihydropyridine 7 with a
1,3-dihalopropane led to the formation of 6-(3'-halopropyl)dihydropyridine
15 and 16. The coupling of the alkyl iodide 16 with the
lithium anion of trimethylsilyl (TMS) hexenylcyanohydrin ether 17 by
nucleophilic substitution provided intermediate 18. Upon acidic and
then basic hydrolysis, the cyanohydrin TMS ether was converted to the
unsaturated diketone 18 in an 82% yield. Although this result is a
linear transformation of 4-methoxypyridine to the required 2,6-disubstituted
2,3-dihydro-4-pyridone, the extra steps to prepare the side chain piece
8 were avoided.
Scheme 4
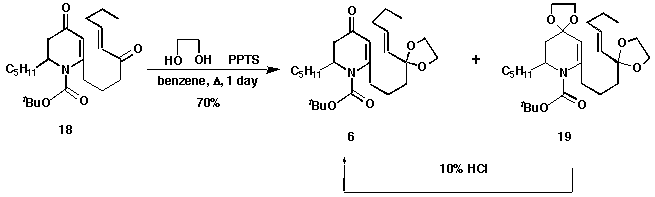
Protection of the keto carbonyl group on the side chain was done by forming
the ethylene glycol ketal 6. The formation of the ketal 6 was
regioselective due to the amide character of the dihydropyridone carbonyl,
which disfavours the ketal formation. The diketal product 19 was formed
as less than 1/3 of the isolated product; however, 19 was found to be
easily converted to the desired 6 upon mild acidic hydrolysis.
Scheme 5
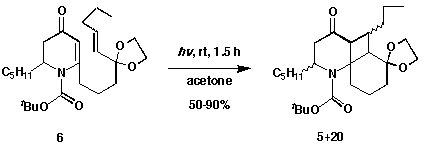
Through this process the substrate for the [2+2] photocycloaddition was
prepared. Irradiation of photosubstrate 6 provided a product mixture of
5 (major) and 20 (minor) in yields up to 90%. From 1H
NMR the ratio of major to minor product was determined to be ca. 6:1.
Because the separation of the two isomers was unsuccessful, the photoadduct
mixture 5 and 20, was used directly in the reductive ring
opening.
Scheme 6
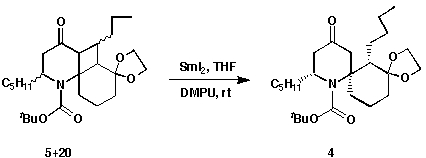
As in earlier model studies,9 the ring opening of the photoadducts
5 and 20 was realized by using SmI2. Only one product was
isolated and purified to give a white solid in 52% yield. The product was
determined to be 4, the ring opened compound derived from the major
isomer of the photocycloadduct from 6. There was no ring-opened product
observed for the minor isomer possibly due to its consumption in the course of
the reduction. Thus, the problem of isomer separation was eliminated. This
reaction is another example of a reductive ring-opening of
a-ketocyclobutane systems with SmI2.9 The reduction
conditions are so mild that the BOC, ketone and ketal groups remained intact.
Scheme 7
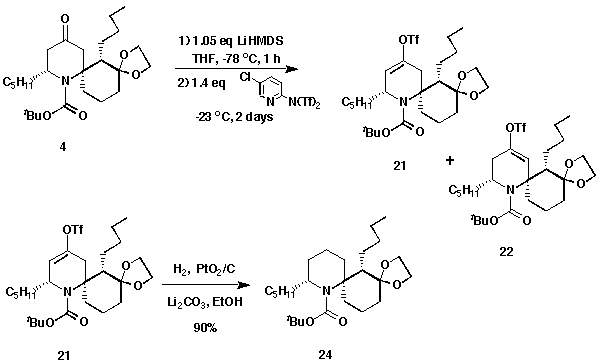
After obtaining 4, which contains the azaspiro skeleton of PHTX,
it was required to reduce and deoxygenate the C-4 keto group. This was
accomplished using Winkler's approach,4a used in his PHTX synthesis.
Generation of the enolate by treating with lithium bis(trimethylsilyl)amide,
and trapping the anion as the vinyl triflate gave a mixture of 21 and
22 in a 90% yield with a 9:1 ratio. The enolate formed was mostly located
at the less hindered a-carbon to the carbonyl group no matter in what
order the base was added. This regioselectivity is beneficial to the synthesis
and the reason will be explained later. Subsequent hydrogenation with PtO2 in
ethyl acetate gave product 24 in 80% yield, with recovery of the
unreacted vinyl triflate isomer 22. It was found that this material
could be reduced only after removal of the N-BOC group. This result
suggests that the vinyl triflate moiety of isomer 22 is hindered by the
nearby ketal group and the tert-butyl group. On removing the BOC group,
it is supposed that the conformational strain was released and the bulky groups
moved away from the vinyl triflate so that the hydrogenation could occur.
Scheme 8
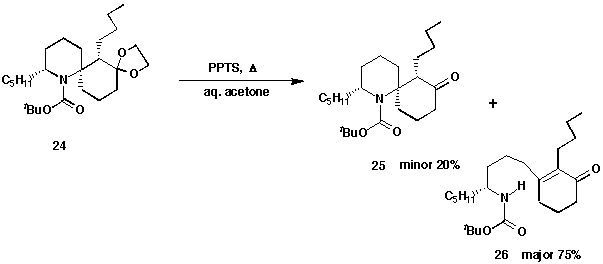
Further manipulations of 24, including removal of the ketal and
N-BOC groups, were attempted. The first attempt involved acid-catalysed
hydrolysis of the dioxolane,10 followed by removal of the BOC
protecting group with trifluoroacetic acid.
Treatment of ketal 24 with a catalytic amount of PPTS in aqueous
acetone under reflux for 3 h produced two products. Unfortunately, the
minor product was shown to be the desired spiro-ketone 25. The major
product was determined to be the ring-opened enone 26, generated through
a subsequent reaction after the ketal was hydrolysed. A pathway for this side
reaction is shown below (Scheme 10).
Scheme 9
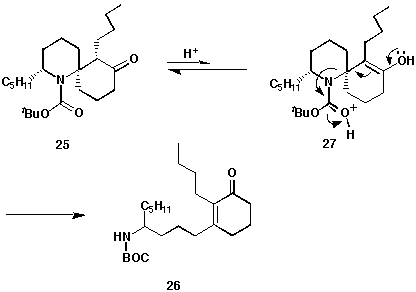
Scheme 10 Proposed pathway for the formation of 26
Further work on improvement of this deprotection to raise the yield of
spiroketone 25 is in progress.
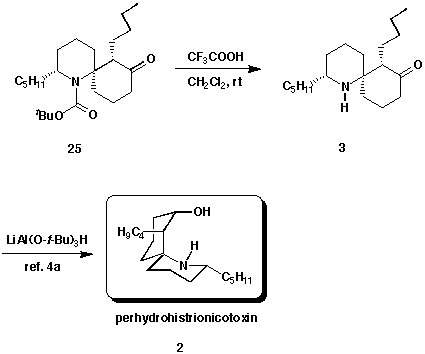
Finally, treatment of 25 with trifluoroacetic acid resulted in the
cleavage of the tert-butyl carbamate and gave amino ketone 3. Since
the amino ketone 3 has been obtained, the final transformation is to
convert the carbonyl group at C-8 to the axial alcohol of 2. This
reaction has been reported by Winkler and coworkers.4a Thus, we
have accomplished a formal synthesis of (+/-)-perhydrohistrionicotoxin 2.
The success of this racemic synthesis will lead to the accomplishment of an
asymmetric synthesis of (-)-perhydrohistrionicotoxin utilizing our asymmetric
synthesis of 2,3-dihydro-4-pyridones.5 Currently, this effort is
underway in our laboratories.
Scheme 11
- Daly, J.W., Karle, I. L., Myers, C. W., Tokuyama, T., Waters, J. A.;
Wiktop, B. Proc. Natl. Acad. Sci. U.S.A., 1971, 68,
1870.
- For reviews: Daly, J. W. and Spande, T. F. in Alkaloids: Chemical and
Biological Perspectives, ed. Pelletier, S. W. Wiley, New York, 1986;
vol. 4, ch. 1, pp. 1-274; Daly, J. W., Garraffo, H. M., and Spande, T. F. in The Alkaloids, ed
Codell, G. A. Academic Press, San Diego, 1993, vol. 43, pp. 185-288.
- For synthetic work and leading references on the histrionicotoxins, see:
(a) Corey, E. J.,
Arnett, J. F., Widiger, G. N. J. Am. Chem. Soc. 1975,
97, 430. (b) Carey, S. C., Aratani, M., Kishi, Y. Tetrahedron
Lett., 1985, 26, 5887. (c) Evans, D. A., Thomas, E. W.,
Cheropeck, R. E. J. Am. Chem. Soc., 1982, 104, 3695. (d)
Venit, J.J., Dipierro M. and Magnus, P. J. Org. Chem., 1989,
54, 4298. (e) Thompson, C.M. , Heterocycles, 1992, 34, 979.
- (a) Winkler, J. D. and Hershberg, P. M. J. Am. Chem. Soc.,
1989, 111, 4852. (b) Stork, G., Zhao, K. J. Am. Chem.
Soc. 1990, 112, 5875. (c) Zhu, J., Royer, J. Quirion,
J.-C. and Husson, H.-P. Tetrahedron Lett., 1991, 32,
2485. (d) Maezaki, N.; Fukuyama, H., Yagi, S., Tanaka, T., Iwata, C. J. Chem. Soc., Chem.
Commun., 1994, 1835.
- Comins, D. L., Joseph, S. P., Goehring, R. R. J. Am. Chem. Soc.,
1994, 116, 4719; Comins, D. L., Goehring, R. R., Joseph, S. P., O'Connor, S. J. Org. Chem.,
1990, 55, 2574 and references cited therein.
- Comins, D. L., Lamunyon, D. H. Tetrahedron Lett., 1989,
30, 5053.
- (a) Babudi, F., Fiandanese, V., Naso, F., Punzi, A. Tetrahedron Lett.,
1994, 35, 2067; (b) Tamao, K., Kodama, S-i; Nakatruka, T. Kiso, Y., Kumada, M. J. Am.
Chem. Soc.,
1975, 97, 4405.
- (a) Hayashi, T., Konishi, M., Kobori, Y., Kumada, M., Higuchi, T., Hirotsu,
K. J. Am. Chem. Soc., 1984, 106, 158. (b) Migani, G.,
Leising, F., Meyrueix, R., Samson, H. Tetrahedron Lett., 1990,
31, 4743.
- Comins, D. L., Zheng, X. J. J. Chem. Soc., Chem. Commun., 1994,
2681.
- Hagiwara, H., Uda, H. J. Chem. Soc., Chem. Commun., 1987,
1351.