| [Molecules: 2] [Related articles/posters: 062 117 101 070 102 ] |

The acid-catalysed decomposition of a-diazocarbonyl compounds is a suitable way to generate other types of electron-deficient species, diazonium ions and carbocations [2]. These transient species are usually intercepted by external nucleophiles to give open-chain products, though the latter in some cases can undergo cyclization to heterocyclic compounds [3]. In a few cases, when a heteroatom is present in the diazo compound itself, nucleophilic cyclization can occur [4]. In our opinion this promising route to heterocyclic compounds appears to have escaped the attention of synthetic chemists, probably because of insufficient information on the mechanism of these processes. In this work we investigate acid-catalysed hydrolysis of two sample diazo ketones: 2,4,6-trimethoxydiazoacetophenone (1) which undergoes efficient cyclization to 4,6-dimethoxy-3(2H)-benzofuranone (2), and diazoacetophenone (3), which gives the intermolecular product, 2-hydroxyacetophenone (4).
Scheme 1
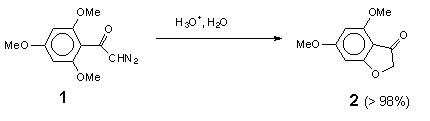
Hydrolysis of the model compound, diazoacetophenone (3), yields 2-hydroxy-acetophenone (4) (Scheme 2) [2].
Scheme 2
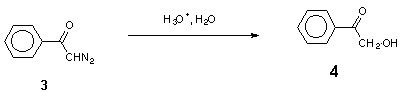
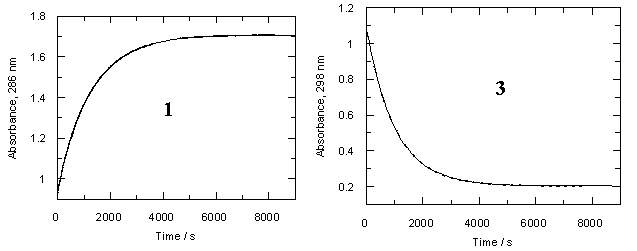
We measured the rates of hydrolysis of both diazocompounds 1 and 3 in dilute aqueous perchloric acid solutions over the concentration range pCH+= 1-4. The decay of diazoketones comformed to the first-order law well, and the observed rates are summarized in Table S1 (for 1) and Table S2 (for 3). The data are displayed as open circles (1) or open triangles (3) in the rate profiles of Figure 2
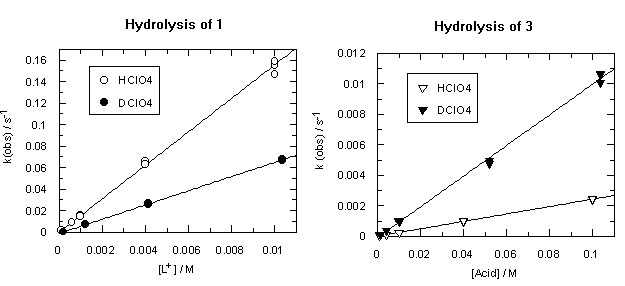
The hydrolysis of both diazocompounds 1 and 3 is an acid-catalysed process, and observed rates of hydrolysis show linear dependence on perchloric acid concentration. Second-order rates calculated from these slopes are shown in Table 1.
| Process | Constant | 1 | 3 |
|---|---|---|---|
| Hydrolysis | kH+ / M-1 s-1 | 15.7 +/- 0.8 | 0.0243 +/- 0.00009 |
| Hydrolysis | kD+ / M-1 s-1 | 6.61 +/- 0.04 | 0.101 +/- 0.001 |
| Hydrolysis | kH+/ kD+ | 2.37 | 0.241 |
| Hydrogen exchangeb | k (H -> D) / M-1s-1 | - | ca. 8 |
| Hydrogen exchangec | k (H -> D) / s-1 | - | (3.4 +/- 0.6) e-005 |
| Hydrolysisc | kobs / s-1 | - | (2.46 +/- 0.01) e-006 |
| Iodinationd | kI- / M-1 s-1 | - | 0.00848+/- 0.00025 |
One can see from Table 1 that trimethoxydiazoacetophenone 1 is over 600 times more reactive than parent 3. Rates of hydrolysis of diazoketones 1 and 3 were also measured in D2O solutions over the same acid concentration range as the H2O data. Observed rates are summarized in Tables S3 and S4 respectively and displayed as filled circles (1) or filled triangles (3) in Figure 2. Rate constants for hydrolysis of 1 and 3 in D2O and solvent isotope effects on these rates are listed in Table 1.
Diazoketones 1 and 3 show a dramatic difference in solvent isotope effect on their rates of hydrolysis (Figure 2). In D2O, decomposition of 1 is substantially slower than in H2O, giving a solvent isotope effect in the normal direction, kH+/kD+ = 2.37. This value and direction of isotope effect is characteristic of rate-determining protonation on carbon [5]. On the other hand, the hydrolysis of diazoacetophenone 3 in D2O is more than four times faster than in H2O; the solvent isotope effect is well below unity, kH+/kD+ = 0.24. Such an inverse solvent isotope effect is a clear sign of pre-equilibrium protonation.
We also found that hydrolysis of diazoacetophenone (3) in D2O is accompanied by deuterium incorporation into it (Scheme 3).
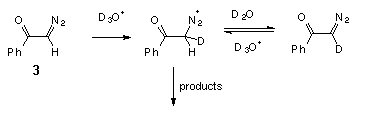
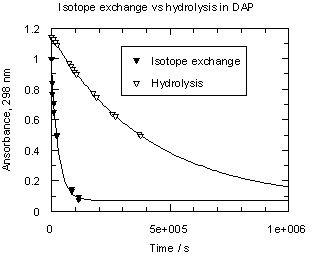
A mass spectroscopic analysis of a recovered sample of the diazo ketone 3, which was treated with a 0.001 M solution of DClO4 in D2O for 5 min (ca. 0.05 hydrolysis tau1/2), showed 90% D-enrichment. This is a clear indication that in the case of the diazoacetophenone (3) pre-equilibrium protonation occurs on carbon. From these data we can also estimate the rate of hydronation of diazoketone 3 as kD+ ca. 8 M-1 s-1. It is interesting to note that this value is very close to the rate of protonation, measured directly, in the case of diazoketone 1.
The addition of iodide ion to the reaction mixtures (ionic strength is still kept at 0.1 M) shows no effect on the rate of decomposition of trimethoxydiazoacetophenone 1; however, it speeds up hydrolysis of 3 (Figure 4).
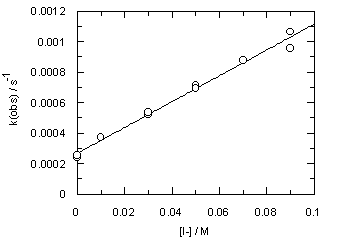
Observed rates are listed in Table S5. Iodide ion catalysis (kI-= 0.00848 +/- 0.00025 M-1 s-1 at [H+] = 0.01 M) indicates that nucleophilic assistance occurs in the rate determining step of diazoacetophenone (3) hydrolysis.
1 Rapid and reversible protonation on oxygen followed by nucleophilic replacement of N2, (Scheme 4).
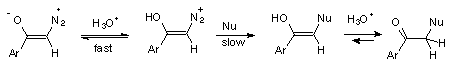
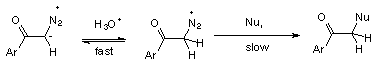
3 Rate determining protonation on carbon followed by fast nucleophilic replacement of N2, (Scheme 6).
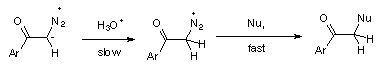
The inverse solvent isotope effect, kH+/kD+ = 0.24, found in the hydrolysis of diazoacetophenone (3), could be accomodated by both mechanisms, 1 and 2. However, hydrolysis of diazoketones by reversible protonation on carbon (mechanism 2) in D2O should be accompanied by hydrogen exchange, while mechanism 1 gives no possibility of such exchange. The D-incorporation into diazoketone 3, accompanying hydrolysis in D2O, supports mechanism 2, indicating that reversible protonation of 3 also occurs on carbon of the diazo group.
The rate-determining loss of nitrogen from protonated diazo ketone 3 can be monomolecular, A1 mechanism, or bimolecular, A2 mechanism, as shown in Scheme 7.
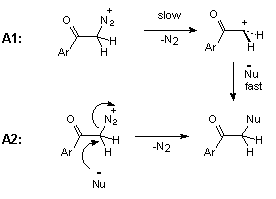
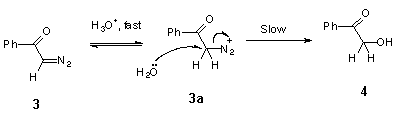
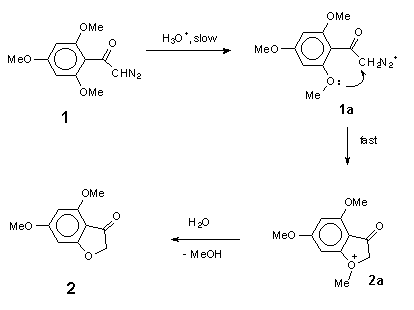
The kinetic data for acid-catalysed cyclization of trimethoxydiazoacetophenone 1 to benzofuranone 2 unfortunately provides no information on steps beyond the rate-determining protonation. However, it is reasonable to expect diazonium ion 1a (Scheme 9), formed in this step, to have a reactivity towards external nucleophiles comparable to that of 3a (Scheme 8), because the diazonium group in ArCOCH2N2+-type ions is not conjugated with the aromatic ring. The fact, that even in the wholly aqueous solutions we found no traces of trapping of diazonium ion 1a by external nucleophile (H2O), indicates that this ion is consumed in some other, much faster process. Most probably, protonation of 1 gives diazonium ion 1a, which then undergoes rapid nucleophilic attack by the oxygen of its ortho-methoxy group to give oxonium ion 2a. Loss of methyl group from 2a in the form of primary carbonium ion CH3+ seems to be very unlikely, and oxonium ion 2a, probably, undergoes hydrolysis by direct displacement to give the final product, 4,6-dimethoxy-3(2H)-benzofuranone 2 (Scheme 9).
This difference in rates and mechanisms between the hydrolysis of 1 and 3 shows that a heteroatom, occupying the right position in a molecule of diazo compound, could be a very effective nucleophile in attacking a protonated diazo group, thus providing a promising approach to heterocyclic synthesis.
5,7-dimethoxy-3(2H)-benzofuranone (2). 40 mg of diazoketone 1 in 8 mL of THF was added dropwise to 30 mL of a stirred solution of aqueous 2 M HCl at room temp. over a period of 5 min. The THF was then removed in vacuum, and the aqueous phase was extracted three times with 20 mL portions of chloroform. The combined organic phases were washed with water, dried over MgSO4, and the solvent was removed to provide 32.5 mg (>98%) of colourless crystals of 2. Mp (without purification!) 139-140oC (lit. 138-140oC [7]). The 1H, 13C, IR and mass spectra of the product are in a good agreement with literature data [7,8].
Diazoacetophenone 3 was prepared by literature procedure [9]. All other materials were best available commercial grades.
Kinetics Rates of hydrolysis of diazoketones 1 and 3 were determined spectrophotometrically by monitoring the changes in absorbance at lambda = 282 and 298 nm respectively. The ionic strength of all solutions was kept at 0.1 M by addition of sodium perchlorate, and the temperature of the reaction mixtures was controlled at 25.0 +/- 0.05oC. Isotope exchange in D2O - (CD3)2SO solutions of 3 was monitored by NMR. Rate constants were calculated by least squares fitting of an exponential function.