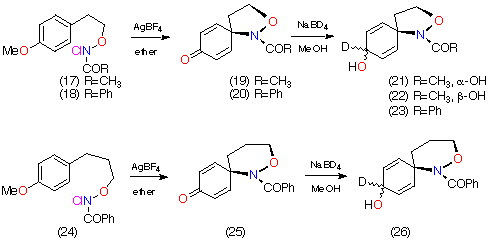
| [Molecules: 2] [Related articles/posters: 062 036 037 107 040 ] |
N-alkoxynitrenium ions are strongly resonance stabilised through the oxygen lone pair. The ¼-bond order is typically 0.9 reflecting significant double-bond character.1 Such stabilisation facilitates their generation from N-alkoxy-N-chloroamides through treatment with silver tetrafluoroborate and other Lewis acids and we have utilised this reaction to synthesise a variety of N-acyl-3,4-dihydro-1H-2,1-benzoxazines 1 and N-acyl-1,3,4,5-tetrahydro-2,1-benzoxazepines 2 (Scheme 1).2 Benzolactams can also be synthesised by this method.3,4
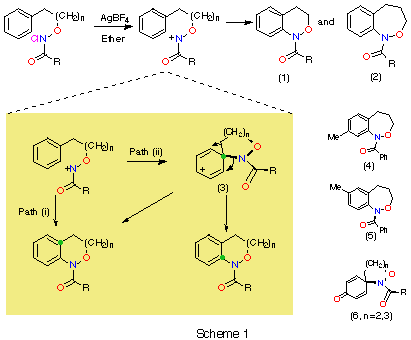
In principle, two mechanisms could lead to the formation of 1 and 2.5 As an alternative to direct ortho attack followed by rearomatisation [pathway (i)], the electrophilic nitrenium ion could react at the ipso position to give 3 [pathway (ii)]. A 1,2-carbon or nitrogen migration would also afford the observed products. We were alerted to the possibility of both mechanisms by the formation of both the 7- and the 8-methylbenzoxazepines 5 and 4 in the ratio 2:1 from the cyclisation of N-chloro-O-3-(p-methylphenyl)propyl benzohydroxamate. In addition, cyclisation of substrates bearing a p-methoxyphenyl group led to ipso attack and formation of the dienones 6.3 Both methyl and methoxy substituents would, however, be expected to activate the ipso position to electrophilic attack.
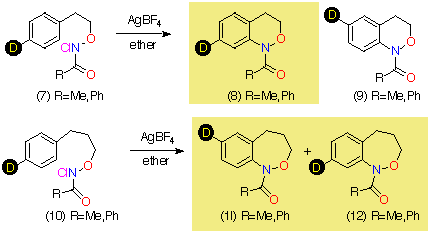
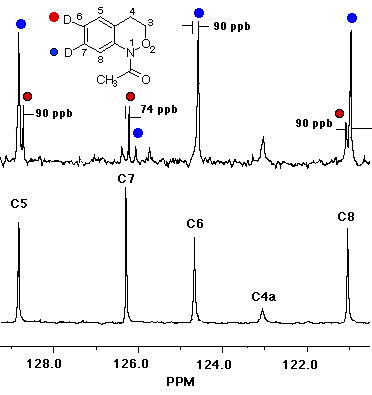
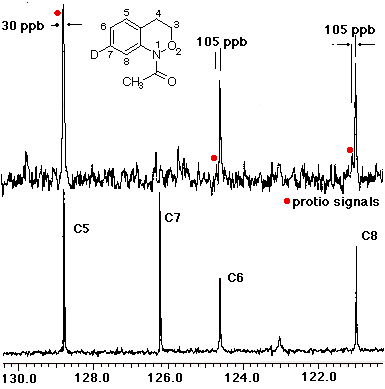
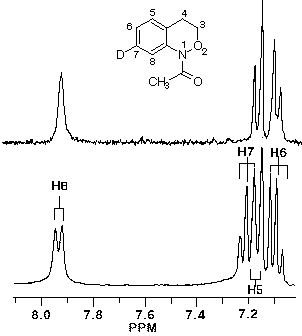
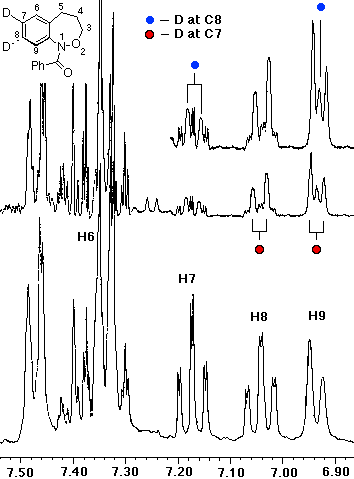
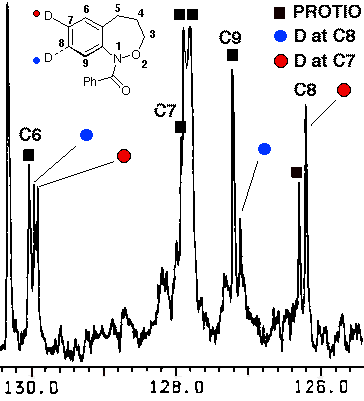
In the case of 7, the sole product was shown by both 1H and 13C NMR to be the 7-deuterio-3,4-dihydro-1H-2,1-benzoxazine 8. 9 was absent in the product mixture. By way of example, in the product from 7 (R=Me), isotope effects in the aromatic region of the 13C NMR spectrum clearly showed that deuterium resided exclusively on the 7-position. This was deduced from the absence of C-7 near d 126, as well as the b isotope shifts of ca. 105 ppb for C-6 (d 124.7) and C-8 (d 121). In addition, H-8, which normlly resonates as a doublet at d 7.93 as a consequence of strong coupling to H-7 in the proton spectrum becomes a singlet in the deuteriated substrate. H-5 and H-6 form a clean AB spin system and H-7 is completely absent. Similar results were obtained for the N-benzoyl analogue.
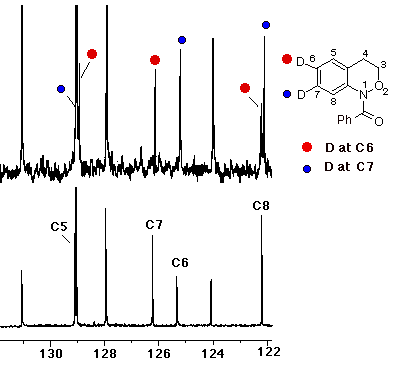
In the case of the formation of the N-benzoylbenzoxazepine ring system from 10 (R=Ph), both the 1H and 13C NMR indicate the presence of mainly the 7-deuterio 11 together with some of the 8-deuterio product 12 indicating that ipso attack followed by 1,2-C migration was the major pathway. The proton spectrum in (CD3)2SO at 370 K (ring and amide conformational isomerism lead to extreme broadening in the aromatic region at room temperature) indicates the presence of the 7-deuterio product through strong doublets at d 6.94 (H-9) and d 7.05 (H-8). A weaker singlet at d 6.94 (H-9) and doublet at d 7.17 (H-8) are diagnostic of the presence of the 8-deuterio species. Some protio material was also regenerated in the cyclisation. In the carbon spectrum the presence of both isomers is indicated by two peaks at C-6 (d 129.6) and C-8 (d 126.2) as well as a major resonance with a b-shift of 99 ppb for C-8 (d 126.25) due to deuterium at C-7 and a minor peak with a b-shift of 95 ppb at C-9 (d 127.1) due to deuterium at C-8 (the shift for C-7 (d 127.5) is masked by the resonances for the benzoyl ring protons)
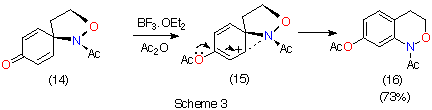
Kikugawa has, however, argued that in competitions between alkoxyamine and carbon migration in dienone-phenol rearrangements, 1,2-N migration is strongly favoured through anchimeric assistance by the nitrogen lone pair; dienone 14 gives almost exclusively the 7-acetate 16 (Scheme 3).6,7 However, such rearrangements are different to those in Scheme 1 as the intermediate cyclohexadienyl cation 15 is resonance stabilised by the ether oxygen of the acetoxy group.
In the case of benzoxazepine formation, while ipso attack appears to be the major pathway, ortho attack to give 12 directly cannot be ruled out.
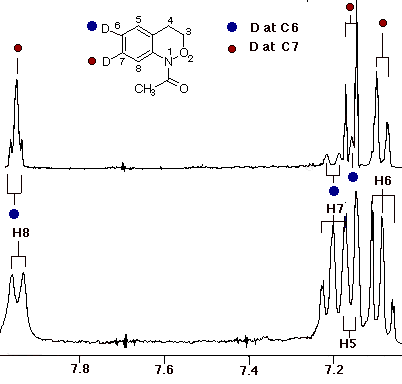
In the resolution enhanced 300 MHz proton spectrum above, the major constituent, the 7-deuteriobenzoxazine (8, R=Me) is evident from the AB pattern for H-6 and H-5, the singlet at H-8 and the greatly reduced signal at H-7. The minor, 6-deuterio isomer (9, R=Me) has a weak doublet for H-7, a singlet (shoulder) for H-5 and a doublet at H-8. In the 13C NMR spectrum below, the 7-deuterio isomer results in 1:1:1 triplet at C-7 and a typical a-deuterium isotope shift of 0.2 ppm. In addition both C-6 and C-8 experience upfield b-isotope shifts of close to 90 ppb while C-5 is largely unaffected. The C-6D isomer results in upfield b isotope shifts for C-5 and C-7 while C-8 is unaffected. The C-7D isomer is clearly the most abundant.
Reduction of N-benzoyl spirocyclohexadienone 20 gave a single isomer which was rearranged with BF3 to give a similar mixture of mainly 7-deuterio-3,4-dihydro-1H-2,1-benzoxazine (M+=240) (8, R=Ph) together with some of the 6-deuterio isomer (9, R=Ph). The carbon spectrum also shows major and minor resonances for C-8 as well as b-shifted signals for C-6 and C-7.
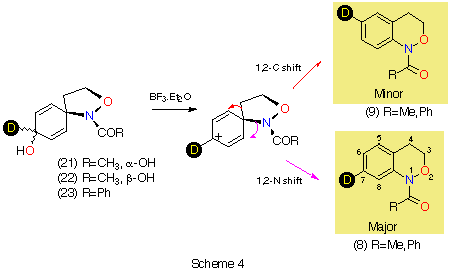
Rearrangement of the dienols 21-23 clearly proceeds through both 1,2-C and 1,2-N migration while nitrogen migration is the predominant process in this case. Neither the stereochemistry of the departing group in 21 and 22 nor the presence of different N-acyl substituents appear to influence the migratory tendencies. We conclude from this that alkoxynitrenium ion cyclisation, which affords only the 7-deuteriobenzoxazine must occur exclusively through direct ortho attack rather than through ipso attack followed by exclusive nitrogen migration.
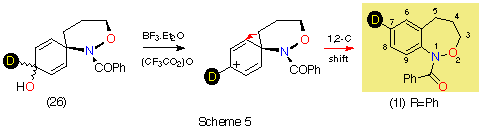
 Reduction
of dienone 25 afforded a mixture of dienols 26 which could not
be separated. The mixture was rearranged by conversion to the trifluoroacetates
in situ and treatment with BF3 The 2,1-benzoxazepine was
isolated by centrifugal chromatography and had a molecular ion of 254 in its
mass spectrum indicating complete retention of deuterium.
Reduction
of dienone 25 afforded a mixture of dienols 26 which could not
be separated. The mixture was rearranged by conversion to the trifluoroacetates
in situ and treatment with BF3 The 2,1-benzoxazepine was
isolated by centrifugal chromatography and had a molecular ion of 254 in its
mass spectrum indicating complete retention of deuterium.
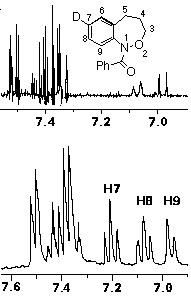
In the resolution enhanced proton spectrum in (CD3)2SO at 370 K (ring and amide conformational isomerism lead to extreme broadening in the aromatic region at room temperature) it can clearly be seen that the sole migration product arises by a 1,2-C shift leading to the 7-deuterio product 11; the triplet for H-7 at d 7.2 is completely absent while H-8 and H-9 form a clean AB spin system. In contrast to the cyclisation process described above where H-9 appears as a singlet and a doublet, no 8-deuterio species is formed in the rearrangement.
In contrast to the five-membered ring, it is clear that the rearrangement is regiospecific in this case. Since cyclisation yields a mixture of 7- and 8-deuterio species, the 8-deuterio isomer 12 must arise through direct cyclisation in that case.
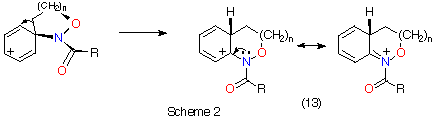
We have argued that the difference in mechanisms arises from the incipient N‹O ¼-bond character. Since the double bond character is endocyclic in the cyclisation process, ipso attack in the formation of benzoxazines would invoke more strain in the transition state (five-membered ring) than in the transition state for ortho attack (six-membered ring).3,5 In the case of benzoxazepine formation, the difference would be less important. AM1 calculations support this assertion; d-H÷ for ipso attack leading to TS2 ( Table 1) is predicted to be larger than that for ortho approach (TS1, Table 1) by 6.3 kcal mol-1 (1 cal = 4.184 J) and the d-S÷ would be expected to be similar for the five- and six-membered ring formation. The difference in the case of the seven-membered ring formation is smaller ( TS6-TS5 = 4.4 kcal mol-1, Table 1) and ortho attack via a seven-membered ring would be characterised by a more unfavourable d-S÷. Thus, rates of cyclisation are most probably similar and both modes of cyclisation would be expected.
AM1 also predicts the relative migratory trends. The barriers to C- and N-migration are predicted to be identical in the formation of benzoxazines from the ipso intermediate (TS3 andTS4, Table 1) and both processes should occur in accordance with what is observed experimentally. In the case of the seven-membered ring formation, the transition state for 1,2-C migration is predicted to be lower in energy than that for 1,2-N migration by 4.6 kcal mol-1 (TS7 and TS8, Table 1).
AM1 also predicts that in each case, the product from carbon migration is thermodynamically more stable in keeping with qualitative predictions (GS4 and GS8 are more stable than GS2 and GS6 by 18 and 16 kcal mol-1, respectively)
| Structure | d-Hf/kcal mol-1 | Structure | d-Hf/kcal mol-1 | ||
|---|---|---|---|---|---|
| GS1 | 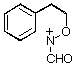
| 209.8 | GS5 | 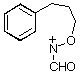 | 203.0 |
| GS2 | 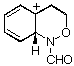 | 190.1 | GS6 | 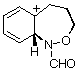 | 180.8 |
| GS3 | 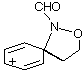 | 203.1 | GS7 | 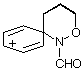 | 191.1 |
| GS4 | 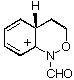 | 171.8 | GS8 | 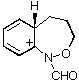 | 164.8 |
| TS1 | 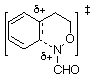 | 217.2 | TS5 | 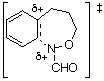 | 209.8 |
| TS2 | 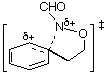 | 223.5 | TS6 | 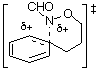 | 214.2 |
| TS3 | 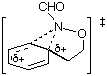 | 221.5 | TS7 | 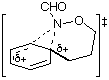 | 211.3 |
| TS4 |  | 221.1 | TS8 |  | 206.7 |
The results presented here clearly suggest that migratory preferences in these systems cannot be generalised. Kikugawa's assertion that N-migration is the preferred mode is valid in the cases he has studied, namely that of the dienone-phenol rearrangements.7 Our results show that even in systems as similar as 23 and 26, migratory aptitudes can be very different indeed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}