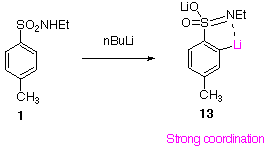| [Molecules: None] [Related articles/posters: 117 037 098 074 105 ] |
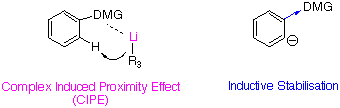
Early work by Hauser and others1 has demonstrated that DoM chemistry of arylsulfonamides allows access to heteroannelation products although this methodology has been sparsely explored, especially on substituted starting materials.3
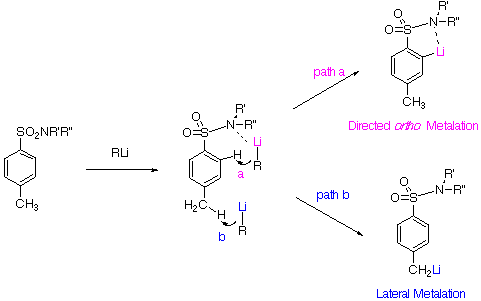
We have investigated DoM (path a) vs. lateral (path b) metalation reactions of tolyl sulfonamides and herein report results concerning kinetic vs. thermodynamic controlled reactions with the aim of improving synthetic utility of these systems.
Under kinetic conditions, both secondary 1 and tertiary 3 p-tolyl sulfonamides undergo ortho deprotonation as a consequence of the strong DMG effect1, to give after deuteriation, products 2 and 4 respectively.. |  |
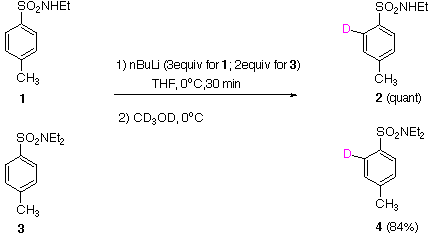
The secondary sulfonamide 1 undergoes thermodynamic ortho deprotonation as evidenced by formation of product 5 in high yield. Even after reflux at 60 oC for 1 h, the derived ortho lithiated species is stable. On the other hand, the ortho lithiated kinetic anion of the tertiary sulfonamide 3, generated at low temperatures, undergoes equilibration at room temp. to the thermodynamically stable benzylic anion which leads to product 6. This undoubtedly occurs via an intermolecular path as already observed in the sulfonate case.4 | |

Using excess LDA, deprotonation of the secondary 1 and tertiary 3 sulfonamides leads, after TMSCl quench, to incomplete formation of products 7 and 6 respectively. If the yield of the conversion 1 to 7 can be improved, a synthetically useful set of conditions for complementary chemoselective deprotonation for the secondary sulfonamide 1 will have been found. | |
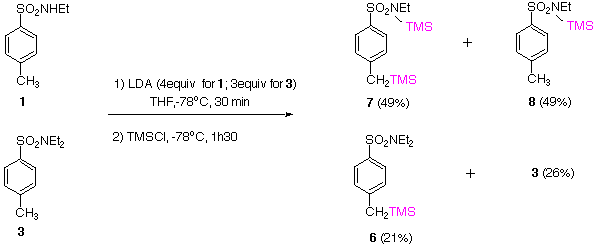
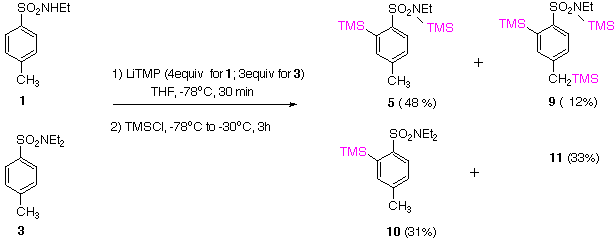
Low temperature deprotonation using LiTMP followed by TMSCl quench on 1 gives mixtures of products 5 and 9 indicating preferred ortho deprotonation. On the other hand, tertiary sulfonamide 3, under identical conditions, affords exclusively product 10 but with starting material (SM) partly recovered. Reactions performed on 1 and 3 with LDA and LiTMP at 0 oC, followed by TMSCl quench, resulted in the formation of a complex mixture of products and decomposition. | |
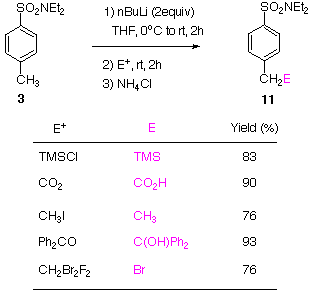
The result of benzylic deprotonation of 3 using BuLi is useful for high yield access of para-functionalized arylsulfonamides 11. The potential for further ortho deprotonation - functionalization of 11, of course with attention to the electrophile (E) introduced, may be recognized. | |
 | Why do we see the DoM reaction with BuLi and LiTMP and not with LDA? |
ortho and lateral metalation pathways for both RLi and LDA may be competitive for several DMGs depending on kinetic or thermodynamic4 reaction conditions as well as presence of complexing additives5 :
Those results reveal the importance of the complex induced proximity effect (CIPE). In the case of LDA metalation, the energetic advantage of complexation to the sulfonamide or amide appears to be greatly reduced compared to BuLi and LiTMP6.
| Why does the ortho lithiated secondary sulfonamide 1 not rearrange at room temperature and above(!) to the more stable benzylic anion 13? |
Perhaps a strongly coordinated, highly aggregated species is formed which is stable to intermolecular rearrangement.