| [Related articles/posters: 005 024 020 ] |
In this contribution, we wish to report on a unique cascade of bond-cleaving and bond-forming reactions which is catalyzed by platinum, a metal which has rarely been used for cascade or domino reactions. The final product obtained from a camphor-derived diyne has some structural elements which also occur in taxol and is therefore an interesting compound for further research.
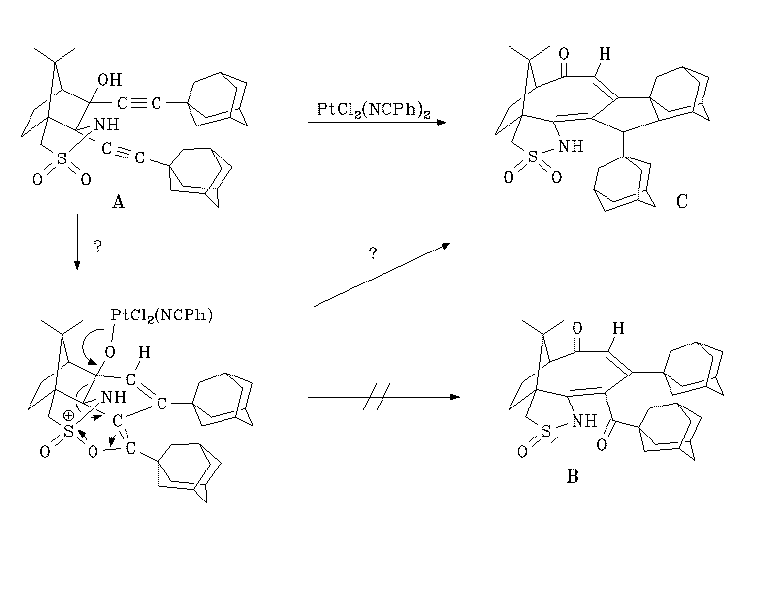
The structural assignment is based on NMR experiments (H-H-COSY, H-C-COSY, ROESY, and long-range C-H correlation (BIRDTRAP)). Although there is much overlap in the 1H NMR spectrum, it is possible to assign most of the signals unequivocally, with the exception of the non-equivalent CH2 and CH groups in the adamantane moieties. We hope to obtain a complete assignment by further NMR experiments. The multiplicities of the NMR signals allow us to exclude the isomer B and assign the structure C to the product.
How can we explain the formation of the product? From experiments with the compound where the adamantyl groups in A are replaced by phenyl groups, we know some details on intermediates. First, the addition of an acid H-X to the diyne leads immediately to an attack of the proton on the triple bond close to the OH group, changing the hybridization of this carbon to sp2 and thus bringing the second carbon atom of the triple bond in close vicinity to the other. This leads to a rapid ring closure to a five-membered carbocyclic ring. The positive charge remaining at the carbon atom not involved in the cyclization is then stabilized by closing a further five-membered ring by forming a bond with an oxygen of the sulfonamide group. We have shown that such stabilized cations are indeed intermediates in what happens further in the phenyl case [8,9] .
In the reaction with PtCl2(PhCN)2 , the required proton for the cyclization is formed by coordination of the platinum to the hydroxy group in A, forming a square planar platinum complex with two chlorides and one benzonitrile as coligands. This can be monitored by NMR by broadening and later disappearance of the OH signal and the detection of free benzonitrile in the solution. In cases where a good nucleophile (e.g. chloride ion) is present in solution, the sulfur-stabilized cation reacts by nucleophilic attack at the carbon atoms of the newly formed double bonds. This leads to the cleavage of the sulfur-oxygen bond, reducing the sulfonamide to the sulfinamide and transferring the oxygen to the carbon atom, thus forming a ketone here [8] . As no good nucleophile is present in the case considered here, the platinum-oxygen bond is finally cleaved, leading to the oxydation of the attached carbon atom to a ketone, and by subsequent electron rearrangement to a cleavage of the C2-C3 bond of the camphor skeleton. The result is a ring enlargement to form a system containing an eight-membered ring which roughly resembles the ring B in the taxol structure. In the case of the compound having phenyl instead of the adamantane groups, the reaction stops at this point. One can show by calculating the energy content of the starting material and the product that the reaction is thermodynamically favoured by ca. 60 - 70 kcal/mol [10] . Alternatively, one can also imagine that the primary attack of platinum is at one of the C-C triple bonds, generating a zwitterionic structure with a negative charge at platinum and a positive charge at carbon, leading to the cyclization of the triple bond system.
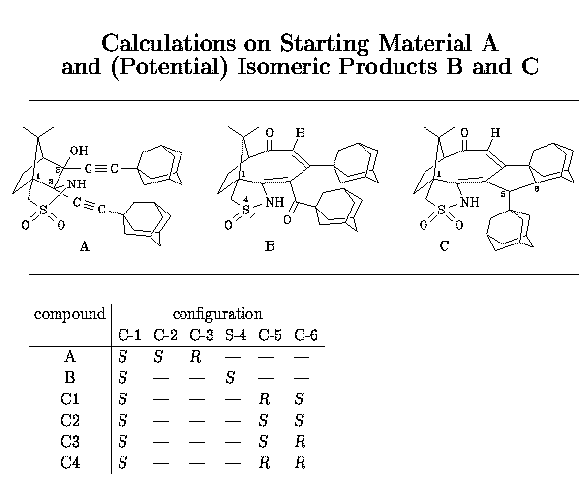
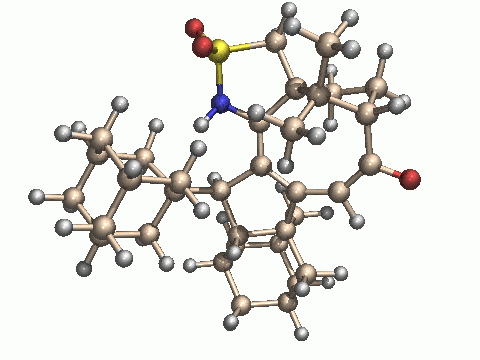
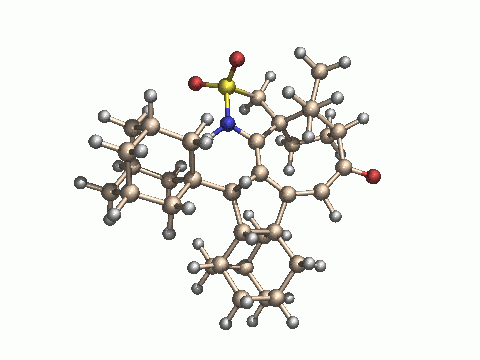
With our adamantane system, we cannot say with certainty whether a sulfur-stabilized cation is also an intermediate here. The broadening and finally disappearance of the OH signal is the only sign of a change of the starting material in the proton NMR spectrum, besides the formation of the final product C. If it were really an intermediate, sulphur reduction and formation of a ketone by transfer of an oxygen atom from sulfur to carbon would be a logical consequence, leading to compound B. From this sulfinamide, however, there would be no way back to the sulfonamide, since the carbonyl group remains far away from sulphur (about 3.4 Ångstrom). In strong contrast to the phenyl case, platinum catalyzes C-H bond activation in one of the adamantanes, probably by insertion into this bond, followed by a C-C bond forming reaction and hydrogen transfer from platinum to carbon. It remains, however, completely speculative in which order these steps occur in the reaction cascade. As no details on the mechanism are known with certainty, we cannot predict which stereoisomer of C is obtained. We have done some semiempirical calculations on the starting materials and potential products to clarify this point (see the calculation part). Based on these results, we show the structures of the most stable isomer isomer C3 and the least stable isomer C4, as optimized by the AM1 method (PM3 gives the same order of stability). The differences in the calculated energies are mainly due to steric effects and ring strain.
In conclusion, we have found a catalytic (turnover of at least 10)
reaction cascade including
{kind=link}
{kind=link}
{kind=link}