| [Related articles/posters: 023 074 079 ] |
We report preliminary results of our study of the mechanism of the Fischer Indole synthesis for a later study on substituted tryptamines. These substituted tryptamines are building blocks for our indoloquinolizine-based potential antitumor compounds.
We draw a comparison between our findings and the new mechanism recently proposed by Kereselidze and discuss the computational evidence that favor one or the other.
To accomplish this we use three Hamiltonians included in MOPAC 6.0 (AM1, MNDO, and PM3). We deal with topics such as the timing of the various protonation steps, and the predictive ability of the three Hamiltonians.
Table of Contents
Fischer indole synthesis is a well established procedure for the synthesis of substituted indoles. Several members of the indole alkaloid familiy, flavopereirin and sempervirin among them, have shown antitumor activity.[1] Since our drug-development efforts focus on the indoloquinolizinium moiety, recently we have become interested in synthesizing substituted tryptamines as intermediates for the synthesis of indoloquinolizinium analogues with enhanced antitumor activity. In order to guide the synthetic process, we started a theoretical study of the mechanism of the Fischer indole synthesis, so we could gain a better understanding of the electronic factors that influence the course of the reaction.
For the reader unfamiliar with the indoloquinolizinium skeleton, the following embedded
structure illustrates the skeleton itself, as well as its basic naming scheme in rings A, B, C
and D. We generated the structure, and its atomic charges, optimizing with PM3. The 2D
rendering is missing a negative charge on the indolic nitrogen and a positive charge on the
pyridinic nitrogen.
| Highlighting | Display options | Highlighting | Display options |
|---|---|---|---|
| Show A ring | Color atoms by charge | ||
| Show B ring | Show electrostatic potential surface | ||
| Show C ring | |||
| Show D ring | Reset |
The Fischer indolization reaction produces ring B of the indoloquinolizinium skeleton. The following image map shows the commonly accepted Robinson mechanism, although we have expanded the proton transfer steps. All the intermediates are linked to their PM3-optimized structures.
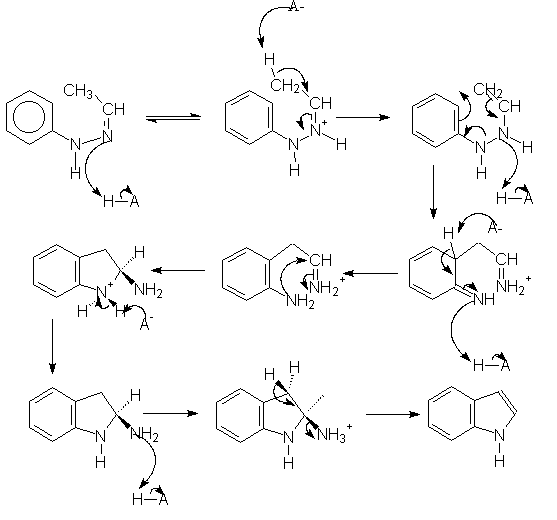
We were unable to find any theoretical studies on the Robinson mechanism, although Kereselidze reported a different mechanism based on semiempirical calculations.[2]. We reproduce the key intermediates of Kereselidze's mechanism, along with their reported energies (in kJ/mol).
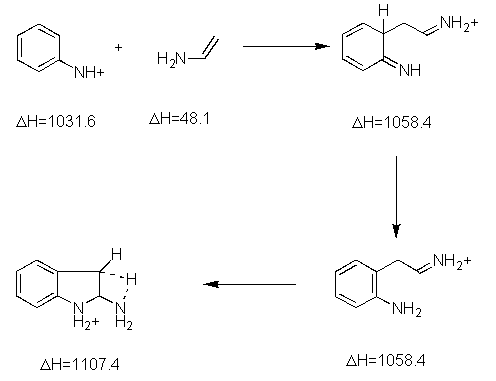
Kereselidze proposes breakup and recombination, although there is no mention of the mechanisn responsible for keeping the pieces together so they can recombine.
All the structures were fully optimized until vibrational analysis showed absence of imaginary frequencies. We started using the Hamiltonians MNDO, AM1 and PM3, but later kept only MNDO and PM3. See the Discussion section for the reasons that prompted us to discard AM1.
We performed all our calculations on MOPAC 6.0 (ported to Windows95 by Victor Lobanov) running on 486 and Pentium PCs. For the (somewhat more demanding) vibrational analysis computations we used a PC equipped with a Pentium II (266 MHz) CPU, 32 MB of RAM and a 4.2 GB hard drive.
The following tables show the heats of formation we obtained using MNDO, AM1 and PM3.
| DHf (kcal/mol) | DHf (kcal/mol) | DHf (kcal/mol) | |
|---|---|---|---|
| AM1 | 59.61 | 221.47 | 73.33 |
| MNDO | 51.55 | 249.60 | 68.92 |
| PM3 | 55.40 | 235.17 | 69.58 |
| AM1 | 201.62 | 174.97 | 178.79 |
| MNDO | 216.11 | 193.54 | 182.87 |
| PM3 | 208.29 | 187.61 | 173.26 |
| AM1 | 31.66 | 182.72 | 55.15 |
| MNDO | 24.16 | 184.16 | 44.15 |
| PM3 | 23.03 | 174.47 | 42.44 |
The table above displays the structures calculated by PM3. The general features are also predicted by the other two Hamiltonians. The changes in energy when going from one intermediate to the next are very similar in all three cases.
The starting arylhydrazone had a conformation folded around the N-N bond, but the optimization yielded an extended conformer. Protonation stabilizes the folded conformer of the hydrazone, but subsequent isomerization to the enamine produces a freely rotating N-N single bond. Protonation of this intermediate and Cope rearrangement produces a non-aromatic species. This aromaticity is recovered in the next step, enabling the nucleophilicity of the nitrogen to form the indolic ring. In this step, only AM1 predicts an increase in energy (which would make sense, given the five-membered ring strain), while MNDO predicts a decrease in energy of 10.67 kcal/mol and PM3 a decrease of 14.35 kcal/mol.
AM1 predicted indole as a transition structure (with only one imaginary frequency). Despite our repeated attempts to guide the structure "downhill" to reach an energy well, the calculation of vibrational frequencies kept showing one imaginary frequency corresponding to the indolic nitrogen. Because of this, we considered prudent to abandon AM1, and continue our work with PM3 and MNDO only.
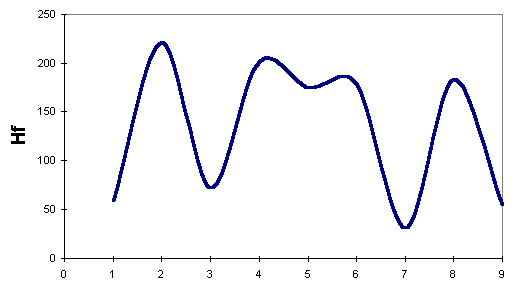
We have plotted the energies for the lowest-energy reaction path, in Robinson's mechanism, in
the following figure:
We have also plotted the energies for Kereselidze's lowest-energy reaction path:
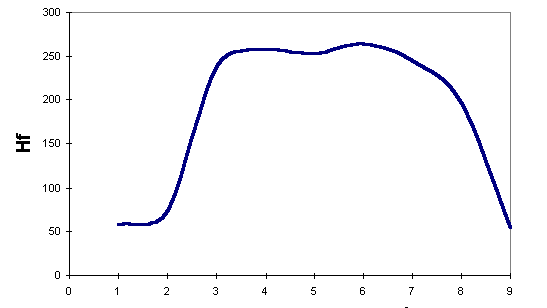
To compare our results against Kereselidze's, we use only those obtained by AM1. Even though we considered AM1 as less suitable for this study, we prefer to compare results obtained using the same Hamiltonian. From the plots, we can see that Robinson's mechanism postulates a minimum- energy path lower in energy than that postulated by Kereselidze.
We found that the three Hamiltonians predict the following intermediate in Robinson's mechanism as more stable than indole itself.
This may help explain why, although the Fischer indole synthesis can produce a variety of substituted indoles, we have not found reports of production of indole by this method. Upon closer examination, we found that all three Hamiltonians predict a pyramidal indolic nitrogen. So it may well be that, in the resulting indole structures, the lone pair on the nitrogen fails to extend the aromatic system to the five-membered ring, and ring strain produces the increased energy.
About the timing of the various protonation steps, the first protonation equilibrium is necessary to activate the isomerization to the enamine, so we have not examined yet the potential energy hypersurface for the unprotonated isomerization. Our calculations show that the Cope rearrangement most likely happens before the protonation of the nitrogen that leaves as ammonia, although to better answer this question we need the results of our transition-state searches for the Cope rearrangement in different stages of protonation.
We can summarize our conclusions as follows:
The authors are grateful to the Consejo Nacional de Ciencia y Tecnología in México (CONACYT) for funding this project through grant 1139P-A9507.