| [Related articles/posters: 051 074 110 ] |
Scheme 1
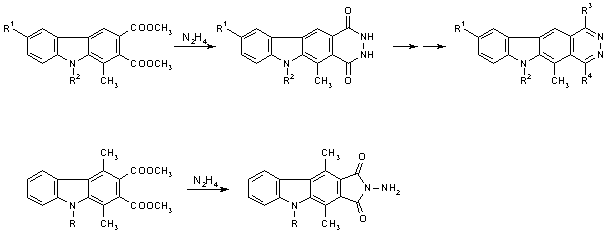
In the case of these compounds as well as of the earlier congeners [5], the tetracyclic skeleton was constructed by reaction of an appropriate 2,3-carbazoledicarboxylic ester with hydrazine hydrate, which gave the carbazole-fused pyridazinedione (see Scheme 1). This approach, however, does not work with 1,4-disubstituted 2,3-carbazoledicarboxylates: here, the corresponding N-aminoimides are obtained instead [7]. We therefore became interested in exploring an alternative pathway to 5,11-disubstituted pyridazino[4,5-b]carbazoles (which feature the same substitution pattern as ellipticine). For this purpose, a cycloaddition reaction between a synthon with a preformed pyridazine ring and another component representing the remaining part of the tetracyclic system was considered a promising strategy.
As an indole-derived diene, an appropriately substituted pyrano[3,4-b]indol-3(9H)-one should be a useful building block, as such compounds had already been employed for the construction of substituted/condensed carbazoles previously, e.g. by Plieninger [8], Moody [9], and Pindur [10]. As the dienophile, we chose the electron-deficient 5-ethanesulfonyl-2-methylpyridazin-3(2H)-one [11], which had been demonstrated to react with 2,3-dimethylbutadiene in a Diels-Alder reaction, affording a phthalazinone derivative [12].
When the dimethyl-substituted pyranoindolone 1 [9] was heated with the pyridazinone 2 [11] in bromobenzene to 156�C, no conversion took place. However, employment of a higher-boiling solvent (1,2,4-trichlorobenzene) was found to effect the cycloaddition reaction at a temperature of 190�C. An excess of 1 was used (with gradual addition), and careful exclusion of air oxygen was found to be crucial. Column chromatography of the reaction mixture afforded the two isomeric cycloaddition products 3a and 3b in a ratio of 1:3 (43% combined yield) as well as two side products (see below). Obviously, compounds 3a,b are formed by spontaneous elimination of carbon dioxide and ethanesulfinic acid from the initially formed, highly strained cycloadducts.
Scheme 2
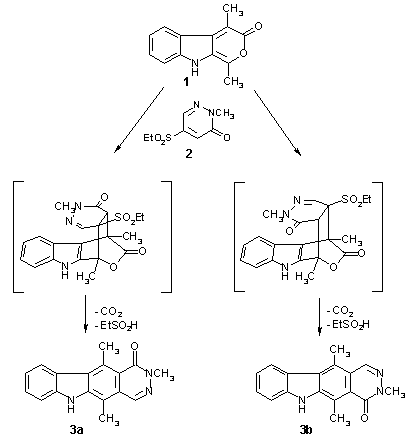
Structural assignment for 3a,b is based on Nuclear Overhauser Enhancement (NOE) difference spectroscopy, using the resonances of the two C-methyl groups, H-10, and/or the pyridazine proton as irradiation points (spectra are available).
In order to examine the generality of this pathway to pyridazino[4,5-b]carbazoles, we also employed the 1-monosubstituted pyrano[3,4-b]indol-3(9H)-one 4 [8] as a diene in the Diels-Alder reaction with the pyridazinone 2. Not unexpectedly, the cycloaddition was found to take place at lower temperature (180�C) as compared to the transformation of 1 into 3a,b, which is explained by the lower degree of steric hindrance at the diene substructure. Interestingly, the product ratio of the resulting carbazole-fused pyridazinones 5a and 5b, which were separated by column chromatography (48% combined yield), was found to be 3:1, thus being reversed with respect to the 3a:3b ratio of 1:3. As the electronic properties of the two pyrones 1 and 4 are almost identical, the reason for this observed reversal in regioselectivity of the cycloaddition must be associated with steric factors. In this context, not only repulsion, but also weak attractive forces between approaching subunits of the reactants should be taken into account, as discussed recently [13].
Scheme 3
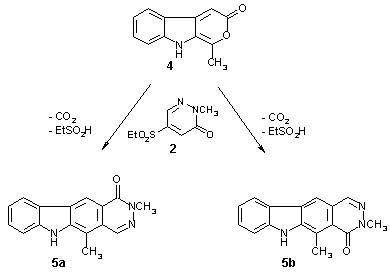
Further experiments, employing 5-ethanesulfonyl-2-benzylpyridazin-3(2H)-one as the dienophile and compounds 1 and 4 as the diene components, gave similar results as described above. Here, the use of a benzyl group as a removable substituent at the pyridazine nitrogen atom should provide a possibility for further transformations of the tetracyclic compounds thus obtained.
We found that also the 1,4-disubstituted pyrano[3,4-b]indol-3(9H)-one 1 undergoes side reactions to some extent under the conditions employed, and we were able to isolate two interesting final products of such a process. These hexacyclic compounds, 6 and 7, both featuring a carbazolocarbazole skeleton, were obtained in 14% and 19% yield, respectively (separation by column chromatography/MPLC). A possible mechanism for the formation of 6 and 7 could involve thermally induced extrusion of carbon dioxide from 1, affording a highly reactive species (A) with a diradicalic or zwitterionic structure. Rearrangement of this intermediate, followed by a sequence of dimerization (B+B for 6, B+C for 7) and oxidation/dehydrogenation finally might lead to the formation of the two isomeric polycycles.
Scheme 4
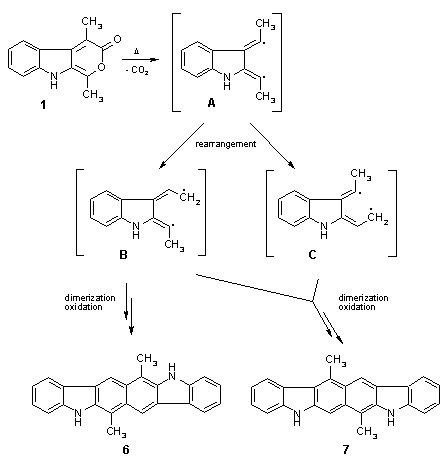
The structures of compounds 6 and 7 were established by a combination of microanalysis, mass and NMR spectroscopy, and in particular by a series of NOE difference experiments which provided full information about the substitution patterns. For compound 7, the key experiment enabled us to assign all four protons at ring A by one single NOE difference spectrum which, in turn, made it possible to determine the position of all other Ar-H, NH, and methyl substructures by means of several consecutive NOE experiments.
Acknowledgement
Support of this work by the "Stiftung Aktion Österreich-Ungarn / Osztrák-Magyar
Akció Alapítvány" is gratefully acknowledged.
NH, 15 June 1998