 ECHET96 Article 081 Iain Coldham
ECHET96 Article 081 Iain Coldham
Re: ECHET96 Article 081 Bob Gawley
Alkylations of N-allyl-2-lithiopyrrolidines. Several analogies to reactions of N-methyl compounds and one surprise
Robert E. Gawley* and Silvio Campagna
Department of Chemistry, University of Miami, Coral Gables, Florida
33124-0431, USA
Introduction and background
The alkylation of a-amino organolithium compounds has been a topic of
interest to synthetic chemists for well over two decades [1-4].
Some recent developments relevant to the present work are Beak's discovery that
N-BOC-pyrrolidine can be enantioselectively deprotonated and alkylated
(eqn. 1), [5,6] and our finding that
2-lithio-N-methyl-pyrrolidines and -piperidines possess exceptional
configurational stability and can be alkylated with a variety of electrophiles
(eqn. 2) [7-9].
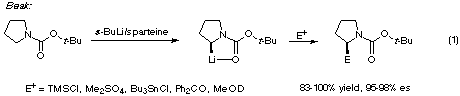
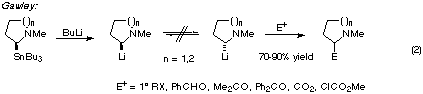
Secondary organolithiums having a heteroatoms such as nitrogen or
oxygen are tetrahedral, and therefore stereogenic. One of the fascinating
aspects of the chemistry of compounds such as these is the stereochemical course
of their reactions with electrophiles. The transition states for SE2
substitutions giving retention and inversion are not very far apart in energy
[10]. Whether the electrophilic substitution (of
lithium by an electophile) occurs with retention or inversion of configuration
at the metalated carbon (or with racemization), sometimes depends on the
electrophile. Early work from the Still group indicated that a-alkoxy
organolithiums react with retention of configuration [11].
However, subsequent studies by Hoppe showed that the lithiated carbamate
illustrated in eqn. (3) affords products of inversion with
acid chlorides, tin chlorides, and the protonating agents triphenylmethane and
acetic acid; reaction with methanol, alkyl halides, esters and anhydrides, on
the other hand, gave products of retention!
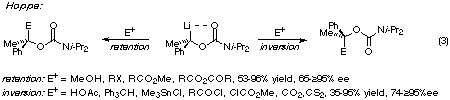
The lithiated N-BOC-pyrrolidines in eqn. (1),
which are configurationally stable at low temperature, give products of
retention of configuration with all electrophiles reported to date
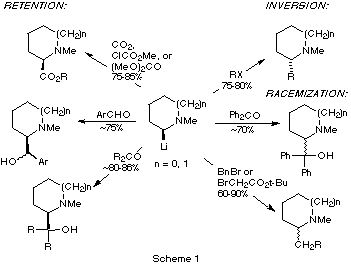
[5,6]. In contrast, 2-lithio-N-methyl-pyrrolidines
and -piperidines afford products with inversion, retention or racemization,
depending on the electrophile (Scheme 1)
[7,9]. For those electrophiles that do not afford
racemic products, the carbonyl-containing electrophiles react with essentially
100% retention for both piperidines and pyrrolidines. With alkyl halides,
piperidines are highly selective, yielding products of inversion with nearly
100% selectivity. Pyrrolidines are somewhat less selective, affording only about
78% inversion of configuration.
One drawback to synthetic applications of some a-hetero-organolithiums
is the fact that they do not always react efficiently with electrophiles. For
example, we have been unsuccessful in achieving high yields of alkylation
products with N-BOC-2-lithio-pyrrolidines and activated (e.g. allylic)
alkyl halides, and unactivated primary alkyl halides are even worse. In
contrast, and for reasons we do not understand, 2-lithio-N-methyl-pyrrolidines
and -piperidines are excellent nucleophiles with a broad spectrum of
electrophiles, affording good yields of products with all electrophiles tested,
regardless of the stereochemistry of the reaction (eqn. 2,
Scheme 1).
In spite of this broad spectrum of reactivity, the processes illustrated in
Scheme 1 have been (so far) limited to N-methyl
compounds. Because we are ultimately interested in applying these reactions to
synthetic problems, we decided to explore the possibility of extending these
processes to heterocycles where there is a removable substituent on nitrogen. To
that end, we tested N-allyl pyrrolidines, and report our preliminary
findings herein. In order to evaluate both chemical reactivity and
stereochemical features, we used educt of high ee (enantiomeric excess). Parenthetically, these are
the same compounds that we used to define the stereochemical features of
[2,3]-anionic and ylide rearrangements of a-aminoorganolithiums
[12].
Off with the BOC, on with the allyl
The procedure for preparing (S)-N-allyl-2-(tributylstannyl)pyrrolidine
is shown in Scheme 2. Beak's (S)-N-BOC-2-tributylstannylpyrrolidine
(92-94% ee, 92:4 - 97:3 er) is treated with B-bromocatechol borane to remove the
BOC group, and immediately alkylated with allyl bromide to afford (S)-N-allyl-2-(tributylstannyl)pyrrolidine,
in 50-55% overall yield after flash chromatography on neutralized silica. This
compound is thermally labile, but can be stored in a freezer under an inert
atmosphere for several days.
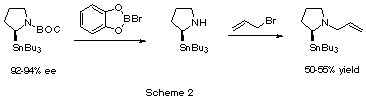
Transmetallations...
In a fashion analogous to the procedures for the N-methyl
compounds, the N-allylpyrrolidinyl stannane can be transmetalated using
butyllithium in THF/TMEDA at low temperature in a few minutes. Although this
organolithium is prone to Wittig rearrangement near room temperature
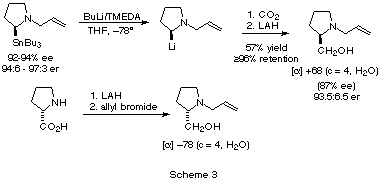
[12], it is stable at low temperature. Quenching the
organolithium with a carbon dioxide stream and reducing the crude N-allylproline
with LAH afforded N-allylprolinol in 57% yield (after flash
chromatography) as a colourless oil. An authentic sample of (S)-N-allylprolinol
was obtained by alkylation of (S)-prolinol. Comparison of the two
specific rotations indicates that the alkylation product is (R), and of
87% ee (93.5:6.5 er). Thus, the reaction of the lithiated N-allylpyrrolidine
occurred with at least 96% retention of configuration. The reaction probably goes
with complete retention, since the differences in specific rotations (of the
beginning stannane as well as the two products) are probably within experimental
error, and because preliminary data reported elsewhere indicate that N-allyl-2-lithiopyrrolidine
is even more resistant to racemization than its N-methyl analogue
[12], which may be due to additional stabilization by
the double bond. These results are summarized in Scheme 3.
Range of electrophiles
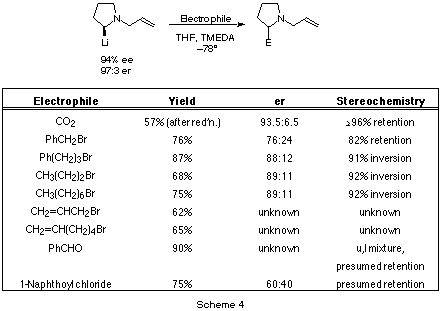
Similarly, a number of other electrophiles were tested, and the results are
shown in Scheme 4. In all cases, the products of alkylation
were formed in excellent yields. The absolute configuration of the products were
confirmed by independent synthesis (E = carbon dioxide, benzyl bromide
[13]), or by order of elution on a Pirkle column
after de-allylation (see below) and acylation with naphthoyl chloride
[14] (E = propyl bromide, heptyl bromide,
3-phenylpropyl bromide). Other configurations are assigned by analogy (E =
PhCHO, naphthoyl chloride). Comparison of the absolute configuration and
enantiomer purity of the various products with that of the stannane reveal that
the trends observed in the N-methyl series (Scheme 1) are largely
followed: electrophiles having a carbonyl add with virtually complete retention
of configuration, while primary alkyl halides react with ca. 90% inversion of
configuration. The 90% stereoselectivity for reactions with alkyl halides is
significantly higher than that observed with 2-lithio-N-methylpyrrolidines,
which were only ca. 78% selective [9] (also showing
inversion).
Never assume you know what will happen....
The surprise is benzyl bromide. In the N-methyl series
(Scheme 1), the product of benzylation was racemic, but
here a 76:24 ratio of enantiomers was found. Even more surprising is that this
alkyl halide reacted with ca. 78% retention of configuration, making it the
only alkyl halide (so far found) that reacts with these lithioheterocycles with
retention!
Allyl group removal
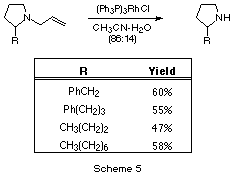
To complete the reaction sequence, we used Wilkinson's catalyst to remove
the allyl group[15] for selected examples, as shown in
Scheme 5. There are some limitations to this protocol: one
is that the allyl group removal fails for N-allylpyrrolidines having a
hydroxyl group in the 2-substituent (i.e. benzaldehyde addition product and
prolinol) and for 1,2-diallylpyrrolidine. The other is that removal of the allyl
group for the compound with the hex-5'-enyl substituent is accompanied by some
double bond migration in the side chain. This latter experiment has only been
done once, however, and efforts to optimize the conditions are in progress.
Summary and conclusions
Replacement of the BOC group in N-BOC-2-tributylstannyl pyrrolidines with
an allyl group, followed by tin-lithium exchange, affords an
a-aminoorganolithium species that reacts with either retention or inversion
of configuration, depending on the electrophile. This two-step sequence may find
use in applications where direct alkylation of the BOC-pyrrolidine is
inefficient, or when the opposite enantiomer is desired.
References
- P. Beak; D. B. Reitz Chem. Rev. 1978,
78, 275.
- P. Beak; W. J. Zajdel; D. B. Reitz Chem. Rev. 1984, 84,
471.
- R. E. Gawley; K. Rein In Comprehensive Organic Synthesis. Selectivity,
Strategy, and Efficiency in Modern Organic Chemistry; ed. B. M. Trost and I.
Fleming, Pergamon, Oxford, 1991; Vol. 3, pp. 65-83.
- T. K. Highsmith; A. I. Meyers In Advances in Heterocyclic Natural
Product Synthesis; ed. W. H. Pearson, JAI, Greenwich, CT, 1991; Vol. 1, pp.
95-135.
- S. T. Kerrick; P. Beak J. Am. Chem. Soc. 1991,
113, 9708.
- P. Beak; S. T. Kerrick; S. Wu; J. Chu J. Am. Chem. Soc. 1994,
116, 3231.
- R. E. Gawley; Q. Zhang J. Am. Chem. Soc. 1993,
115, 7515.
- R. E. Gawley; Q. Zhang Tetrahedron 1994, 50,
6077.
- R. E. Gawley; Q. Zhang J. Org. Chem. 1995,
60, 5763.
- E. D. Jemmis; J. Chandrasekhar; P. v. R. Schleyer J.
Am. Chem. Soc.
1979, 101, 527.
- W. C. Still; C. Sreekumar J. Am. Chem. Soc. 1980,
102, 1201.
- R. E. Gawley; Q. Zhang; S. Campagna J. Am. Chem. Soc.
1995,
117, 11817.
- J. E. Nordlander; M. J. Payne; F. G. Njoroge; M. A. Balk;
G. D. Laikos; V. M. Vishwanath J. Org. Chem. 1984, 49,
4107.
- W. H. Pirkle; C. J. Welch; G. S. Mahler; A. I. Meyers; L.
M. Fuentes; M. Boes J. Org. Chem. 1984, 49, 2504.
- C. Bennett; B. Leguzza; B. Ganem Tetrahedron Lett.
1981,
22, 1483.