| |
[ Molecules: 19 ] [ Related articles/posters : 062 103 046 036 102 ] |
Department of Chemistry, University of New England, Armidale, New South Wales 2351, Australia
The NMR spectra of these compounds are characterised by an unusual degree of line broadening which we have discovered is attributable to two processes; slow E/Z-isomerism about the amide bond and conformational changes in the oxazepine ring.
N-benzoyl-1,3,4,5-tetrahydro-2,1-benzoxazepine was the first member of this group to be identified. An X-ray structure indicated a chair conformation for the aliphatic ring with the phenyl substituent folded back over the aromatic portion of the benzoxazepine skeleton (ring and carbonyl oxygens cis. AM1 calculations give a similar lowest energy structure.
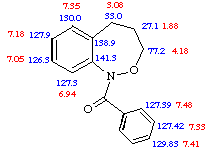
The 1H NMR spectrum showed extreme line broadening for almost all signals in the aliphatic and aromatic regions. Raising the temperature to 375K in D6-DMSO resulted in sharpening of all resonances and the aromatic region was readily assigned as shown. Line broadening at room temperature was therefore particularly evident in the o-protons of the benzoyl ring as well as H8 and H9 on the benzoxazepine skeleton. Full 1H and 13C NMR assignments in D6-DMSO are shown below.
In contrast to theN-benzoyl derivatives,N-pivaloylbenzoxazepine (5) exhibited sharp resonances in both the aliphatic and aromatic regions and both proton and carbon spectra indicated the presence of only one isomer. E/Z isomerisation must be significantly slower in this case and the equilibrium is dominated by one isomer, presumably due to the bulk of thetert-butyl group.
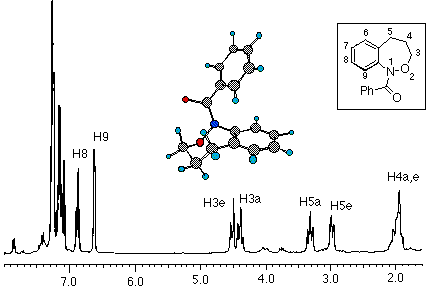
N-acetylbenzoxazepine (4) also exhibited extensive line broadening in its room temperature 1H NMR spectra in CDCl3 as well as the presence of major and minor isomers in the 13C and 1H spectra. Room temperature spectra were therefore recorded in the slow exchange region. Rigorous analysis of the effects of elevated temperature on the aliphatic regions of the proton and carbon spectra ofN-acetylbenzoxazepine (4) in CDCl3afforded coalescence data for the isomerisation process (Table 1)
| Position | Spectrum | Dfreq./Hz | Tc/K | kf/s -1 | kb/s -1 | DG ýkcal mol -1 |
|---|---|---|---|---|---|---|
| Me | 1H | 119 | 325 | 113.3 | 239 | 14.8 |
| Me | 13C | 102.5 | 324 | 99.1 | 205.9 | 14.9 |
| C3 | 13C | 124.3 | 327 | 120.4 | 250.0 | 14.9 |
| C4 | 13C | 19.6 | 308 | 19.0 | 39.4 | 15.1 |
In the slow exchange region, the proton integrals gave the relative proportions of the two isomers as 1:0.65. Detailed analysis afforded forward and reverse rate constants at each of the four coalescence temperatures from which EA fand EA bwere calculated to be 20.1 and 20.2kcal mol -1respectively. Approximating equal populations, DG ýwas calculated to be between 14.8 and 15.1 kcal mol -1(Table 1)
Like (4),N-2-methylpropanoylbenzoxazepine (6) exhibited two amide isomers at room temperature and all proton resonances were exceedingly broad ( 1H NMR spectrum at r.t. ).
In contrast to theN-benzoyl,N-acetyl and N-2-methylpropanoylbenzoxazepine derivatives above,N-acetylnaphthoxazepine (9) exhibited no line broadening in its 1H spectrum and both the proton and carbon spectra indicated the presence of two amide isomers. Elevation of the temperature in D6-DMSO resulted in time averaged environments for all aliphatic and aromatic resonances as a result of rapidE/Zisomerisation.
N-benzenesulfonylbenzoxazepine (7) exhibited no line broadening in the aromatic region of its 1H NMR spectrum/link. In addition, the carbon spectrum indicated one set of resonances. TheN�Sbond in sulfonamides is longer than theN�Cbond in amides. In addition, the barrier to rotation aboutN�Sbonds is much smaller than that found for the analogous amides. Thus, the absence of broadening is most probably due to averaging of the chemical environments. A NOESY spectrum also indicated correlations from the benzenesulfonylorthoprotons to both the C9�H and the methylene protons adjacent to the ring oxygen.
It is clear that the slow isomerism at these temperatures involves flipping between energetically identical chair conformations similar to that observed in the solid state or predicted by AM1 calculations.
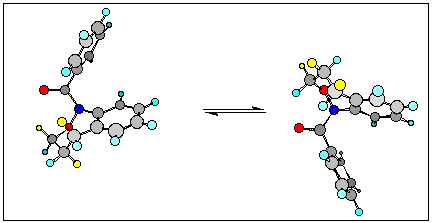
The anisotropic shielding of aromatic protons at C8 and C9 adjacent to nitrogen in the benzoxazepine ring confirm that the oxygens arecis.
The pivaloyl substrate (5), in which only one isomer was prevalent at room temperature, also froze to a chair conformation below 250K as did the benzenesufonyl derivative (7) and in the latter case there was no evidence for slowing of isomerisation about theN�Sbond although chemically distinct methylene protons were broader than in the case of the benzoyl substrates.
Low temperature 1H NMR spectra of acetylbenzoxazepine (4) and 2-methylpropanoylbenzoxazepine (6) indicated the presence of chair conformers for both theEandZ-isomers. A spectrum of the acetyl compound (4) at 220K diplayed overlapping equatorial and axial benzylic resonances at d2.85 and d3.15 and in one isomer the methylene hydrogens adjacent to oxygen resonate normally at d4.2 (axial) and d4.4 (equatorial). The same protons in the other amide isomer also overlap at ~ d4.4.
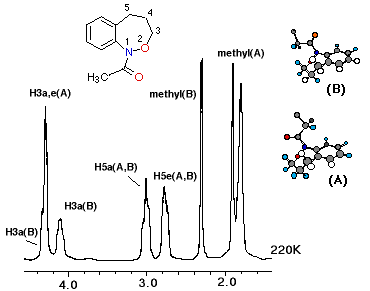
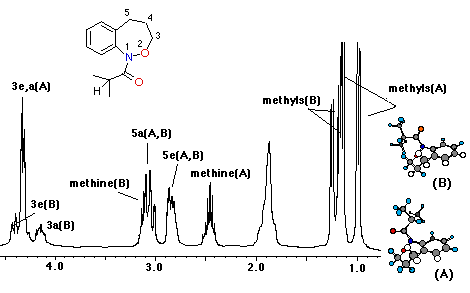
The unsubstituted benzoxazepine (8) exhibited a chair conformation at lower temperatures than the other benzoxazepines and at 220K, only the benzyl protons were clearly resolved into distinctly different environments. Isomerisation in this substratewould be expected to be a faster process when compared toN-acylated derivatives.
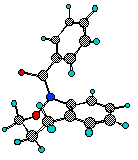
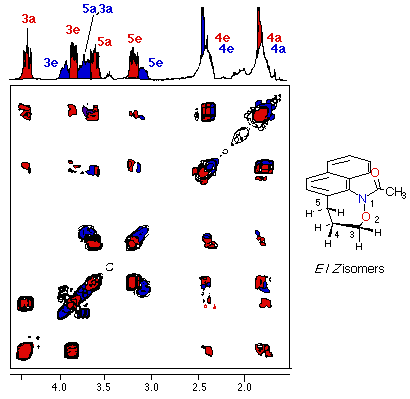
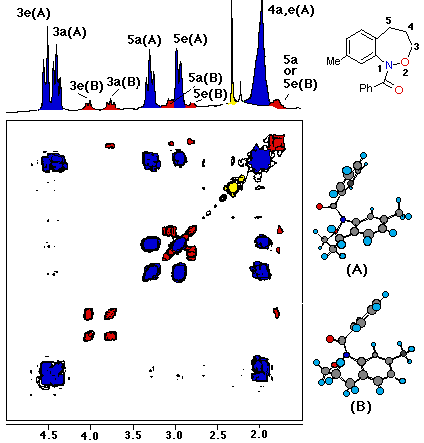
Each of theEandZ-isomers ofN-acetylnaphthoxazepine (9) was conformationally stable at room temperature. Axial and equatorial benzylic and oxymethylenic hydrogens are clearly discernable for each isomer. Resonances for the major isomer (red) and minor isomer (blue) are depicted in the accompanying COSY spectrum of the aliphatic region.
The benzylic protons of the unsubstituted parent (8), which were the only methylene clearly resolved at low temperature, coalesced at 242K yielding a rate constant of 216.2 s -1indicating the fastest isomerisation of all the chair forms studied. The DG ýof 10.98 kcal mol -1for the ring inversion was correspondingly the smallest that we observed. Most typically, the DG ýwere in the region of 11.5-12.5 kcal mol -1and the reduced value for the unsubstituted benzoxazepine indicates that a strong contributor to free energy barrier to inversion is steric hindrance between the acyl substituent and the aromatic proton on the 9�position. In the case of the acetyl (4) and 2-methylpropanoyl (6) substrates, one isomer has an inversion barrier in the above range but the other has a measurably higher barrier to inversion. In the case of the isobutanoyl substrate, coalescence of one set of the benzylic protons ( DG ý=13.6 kcal mol -1) and the isopropyl methyls ( DG ý=14.0 kcal mol -1) are found for the isomer in which the amide and ring oxygens arecisie the isopropyl group over the aromatic ring (6B, Table 2). Similarly, one isomer of the acetyl substrate (4) has a higher barrier of 12.4 kcal mol -1and most probably corresponds to the isomer with methyl over the aromatic ring ie the methyl upfield (4B, Table 2). Thus, spacially demanding groups on theN�acyl substituent measurably slow the rate of ring inversion and particularly so if the acyl sidechain is trans to the ring oxygen. The extreme case of this is found for the naphthoxazepine where ring mobility is very slow, even at room temperature.
| Benzoxazepine | Coalescence | DFreq./Hz | J/Hz | Tc/K | DG ý/kcal mol -1 | kc/s -1 |
|---|---|---|---|---|---|---|
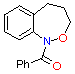 (1) (1)
| benzylic CH2 O�CH2 O�CH2Tw-bt | 103.0 38.0 81.3 | 13.2 12.2 - | 274 260 247 | 12.42 12.19 11.30 | 238.9 107.4 180.6 |
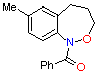 (2) (2)
| benzylic O�CH2 O�CH2Tw-bt | 112.3 40.8 78.3 | 13.44 11.74 - | 283 265 247 | 12.82 12.42 11.32 | 259.9 110.8 173.9 |
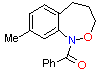 (3) (3)
| benzylic O�CH2Chair O�CH2Tw-bt | 102.2 39.1 80.6 | 13.23 11.74 11.31 | 275 260 245 | 12.49 12.19 11.19 | 238.2 107.7 189.4 |
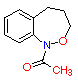 (4) A (4) A
| benzylic O�CH2 | 72 63.2 | 12.6 11.0 | 255 253 | 11.71 11.68 | 174.0 152.6 |
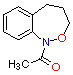 (4) B (4) B
| benzylic | 72.0 | 12.6 | 270 | 12.42 | 174.0 |
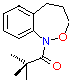 (5) (5)
| benzylic O�CH2 | 103.0 34.0 | 12.6 11.5 | 254 241 | 11.50 11.31 | 238.9 98.1 |
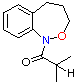 (6) A (6) A
| benzylic O�CH2 methyls | 68.0 80.0 21.0 | 14.5 12.38 - | 256 254 240 | 11.76 11.61 11.62 | 170.5 190.1 46.7 |
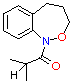 (6) B (6) B
| benzylic methyls | 63 47.3 | 15.2 - | 294 297 | 13.62 14.02 | 162.4 105.1 |
 (7) (7)
| benzylic O�CH2 | 144.0 62.0 | - - | 270 260 | 12.10 12.07 | 319.9 137.7 |
 (8) (8)
| benzylic | 97.3 | 13.87 | 242 | 10.98 | 216.2 |
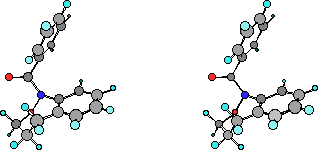
All low temperatures NMR shifts and coupling data accord with a chair conformation and isomerism involves interchange between two such forms, a process that interchanges protons between equatorial and axial environments. As outlined above, this is the structure in the solid state as well as that predicted by semiempirical AM1 calculations. Table 3 below gives AM1 optimised energies and geometries in rotatable form for various conformations of both parent benzoxazepine (8) andN-benzoylbenzoxazepine (1). In both cases the chair form is certainly the lowest energy structure. A search of the geometrical surface located two other local minima higher in energy than this chair form. These are the boat and the twist-boat forms. In both cases, the boat conformation turned out to be the highest in energy although the difference between boat and twist-boat forms for (1) was small whereas for the parent benzoxazepine (8) the twist-boat was only marginally higher in energy than the chair form.
| AM1 Geometries Benzoylbenzoxazepine (1) | DHf | AM1 Geometry Benzoxazepine (8) | DHf |
|---|---|---|---|
(click to change display and rotate) | Chair 24.83 | (click to change display and rotate) | Chair 13.36 |
(click to change display and rotate) | Twist-boat 27.06 | (click to change display and rotate) | Twist-boat 13.90 |
(click to change display and rotate) | Boat 27.83 | (click to change display and rotate) | Boat 18.39 |
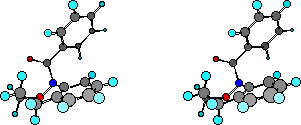
In the low temperature NMR spectra of benzoyl derivatives (1)-(3), and particularly in the 8-methylderivative (3), a second low temperature conformer is evident. A low temperature COSY spectrum at 220K is shown below and satellite resonances for axial and equatorial hydrogens of the oxymethylene ( d3.75 and d4.05) as well as the benzylic methylenes ( d3.05 and d2.80) are evident and correlated through the methylene at C4.
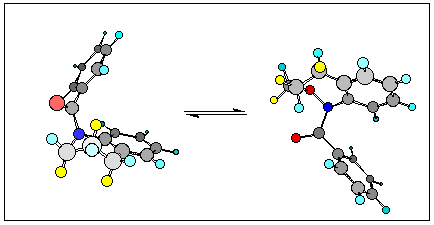
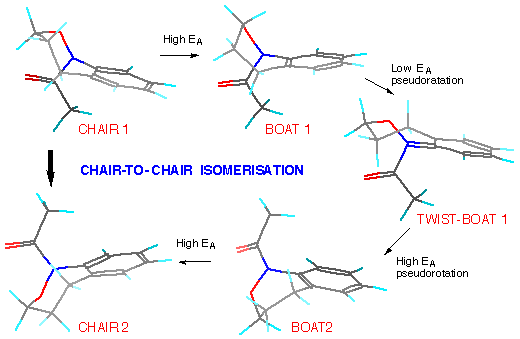
Models indicate that a twist-boat to twist-boat interconversion can proceed through a pseudo-rotation via one boat conformation.
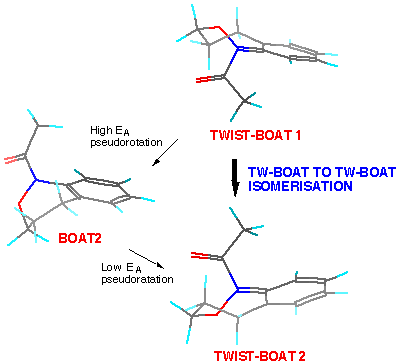
Chair-to-chair interconversion involves a comparatively strained flipping to the nearest boat conformer (but with minimal change at nitrogen), a high EApseudo-rotation via one twist-boat form to the alternative boat followed by flipping once again.
References