| [Molecules: 6] [Related articles/posters: 060 063 024 069 009 ] |


![]() The reactivity of ketenimines
(R1-N=C=CR2R3) has been extensively studied
since their original preparation by Staudinger in 1921.1 The
addition of different nucleophiles to the central sp carbon and the
cycloaddition reactions to produce heterocyclic compounds have received
considerable attention in the literature dealing with ketenimines.2
Concerning their cycloaddition reactions, just two papers describing briefly
the intermolecular cycloaddition of ketenimines with imines have been
reported.3
The reactivity of ketenimines
(R1-N=C=CR2R3) has been extensively studied
since their original preparation by Staudinger in 1921.1 The
addition of different nucleophiles to the central sp carbon and the
cycloaddition reactions to produce heterocyclic compounds have received
considerable attention in the literature dealing with ketenimines.2
Concerning their cycloaddition reactions, just two papers describing briefly
the intermolecular cycloaddition of ketenimines with imines have been
reported.3
![]() In this poster we describe representative examples of the previously unreported
intramolecular [2 + 2] cycloaddition imine-ketenimine.
In this poster we describe representative examples of the previously unreported
intramolecular [2 + 2] cycloaddition imine-ketenimine.
![]() For this purpose we have prepared N-[2-(alkylideneaminomethyl)phenyl]
diphenyl, trimethylsilyl and methylphenylketenimines. For the preparation of
the imine fragment aromatic or aliphatic aldehydes and acetophenones have been
used. The ketenimine fragment was furtherly generated by reaction of
iminophosphoranes with ketenes.
For this purpose we have prepared N-[2-(alkylideneaminomethyl)phenyl]
diphenyl, trimethylsilyl and methylphenylketenimines. For the preparation of
the imine fragment aromatic or aliphatic aldehydes and acetophenones have been
used. The ketenimine fragment was furtherly generated by reaction of
iminophosphoranes with ketenes.
![]() Results and discussion
Results and discussion
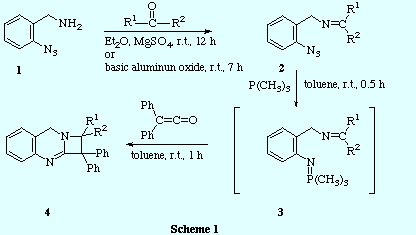
![]() The reaction of o-azidobenzylamine 1 with several carbonyl
compounds gave the corresponding imines 2. Then, the thus obtained
azido-imines were treated with trimethylphosphine to generate the
trimethyliminophosphoranes 3 which were not isolated but when treated with diphenylketene, yielded the corresponding derivatives of the new heterocyclic system
azeto[2,1-b]quinazoline 4
The reaction of o-azidobenzylamine 1 with several carbonyl
compounds gave the corresponding imines 2. Then, the thus obtained
azido-imines were treated with trimethylphosphine to generate the
trimethyliminophosphoranes 3 which were not isolated but when treated with diphenylketene, yielded the corresponding derivatives of the new heterocyclic system
azeto[2,1-b]quinazoline 4
![]() The use of different carbonyl compounds (aldehydes, ketones) allowed us to
introduce a variety of substituents at the stereogenic centre in the chiral
1,1-diphenylazetoquinazolines 4
The use of different carbonyl compounds (aldehydes, ketones) allowed us to
introduce a variety of substituents at the stereogenic centre in the chiral
1,1-diphenylazetoquinazolines 4
| Azeto[2,1-b]quinazolines 4. | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | (%) | ||
| 4a | H | CH(CH3)2 | 36 | ||
| 4b | H | E-CH=CH-C6H5 | 72 | ||
| 4c | H | 3-furyl | 50 | ||
| 4d | H | 4-CH3O-C6H4 | 98 | ||
| 4e | H | 4-NO2-C6H4 | 81 | ||
| 4f | CH3 | 4-NO2-C6H4 | 46 | ||
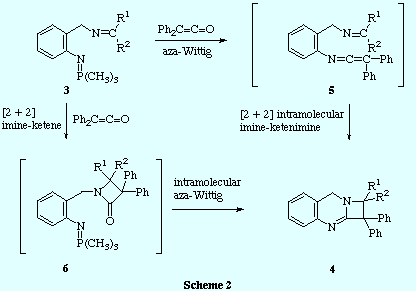
![]() Two mechanistic pathways can be proposed to explain the formation of compounds
4. The first one involves an initial aza-Wittig reaction between the
iminophosphorane function and the ketene to give a imino-ketenimine
intermediate 5 that undergoes intramolecular [2 + 2] cycloaddition. It
is well known that ketenes reacts with imines to produce 2-azetidinones by a [2
+ 2] cycloaddition. Taking this into account, a second mechanism may be
envisaged: the ketene reacts in a [2 + 2] intermolecular cycloaddition with the
imine fragment of the compound 3 to give a 2-azetidinone 6 which
further experiences an intramolecular aza-Wittig reaction between the
iminophosphorane function and the carbonyl group of the 2-azetidinone ring to
give the azetoquinazoline 4.
Two mechanistic pathways can be proposed to explain the formation of compounds
4. The first one involves an initial aza-Wittig reaction between the
iminophosphorane function and the ketene to give a imino-ketenimine
intermediate 5 that undergoes intramolecular [2 + 2] cycloaddition. It
is well known that ketenes reacts with imines to produce 2-azetidinones by a [2
+ 2] cycloaddition. Taking this into account, a second mechanism may be
envisaged: the ketene reacts in a [2 + 2] intermolecular cycloaddition with the
imine fragment of the compound 3 to give a 2-azetidinone 6 which
further experiences an intramolecular aza-Wittig reaction between the
iminophosphorane function and the carbonyl group of the 2-azetidinone ring to
give the azetoquinazoline 4.
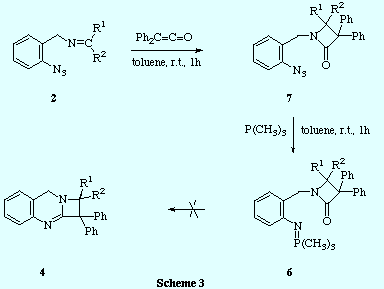
![]() We have prepared the 2-azetidinone-iminophosphorane 6 by an unequivocal
route and proved that this compound did not undergo intramolecular aza-Wittig
reaction to produce the azetoquinazoline 4, even at high temperatures, and so the second mechanism mentioned above
for the formation of 4 can be disregarded.
We have prepared the 2-azetidinone-iminophosphorane 6 by an unequivocal
route and proved that this compound did not undergo intramolecular aza-Wittig
reaction to produce the azetoquinazoline 4, even at high temperatures, and so the second mechanism mentioned above
for the formation of 4 can be disregarded.
![]() Although ketenimines show a very strong absorption at ca. 2000 cm-1
in their IR spectra,we could not observe any absorption in
this region when a IR spectrum of the reaction mixture was registered
inmediately after the addition of diphenylketene to the solution of
iminophosphorane 3;it seemed that the intramolecular [2 + 2]
cycloaddition imine-ketenimine took place immediately following the formation
of the imino-ketenimine.
Although ketenimines show a very strong absorption at ca. 2000 cm-1
in their IR spectra,we could not observe any absorption in
this region when a IR spectrum of the reaction mixture was registered
inmediately after the addition of diphenylketene to the solution of
iminophosphorane 3;it seemed that the intramolecular [2 + 2]
cycloaddition imine-ketenimine took place immediately following the formation
of the imino-ketenimine.
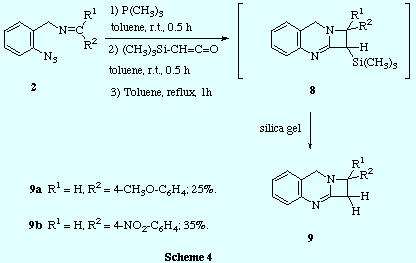
![]() In contrast, the reaction of trimethyliminophosphoranes 3 with
trimethylsilylketene led to a ketenimine intermediate that could be evidenced by
IR spectroscopy (strong absorption at 2004 cm-1) and which was
persistent at room temperature for several hours. The intramolecular
cycloaddition was notably accelerated by heating, and we could not
isolate from the reaction mixture the expected C1-trimethylsilyl
azetoquinazoline 8, but instead, after purification by silica gel column
chromatography, we obtained directly the C1 unsubstituted azetoquinazoline 9
In contrast, the reaction of trimethyliminophosphoranes 3 with
trimethylsilylketene led to a ketenimine intermediate that could be evidenced by
IR spectroscopy (strong absorption at 2004 cm-1) and which was
persistent at room temperature for several hours. The intramolecular
cycloaddition was notably accelerated by heating, and we could not
isolate from the reaction mixture the expected C1-trimethylsilyl
azetoquinazoline 8, but instead, after purification by silica gel column
chromatography, we obtained directly the C1 unsubstituted azetoquinazoline 9
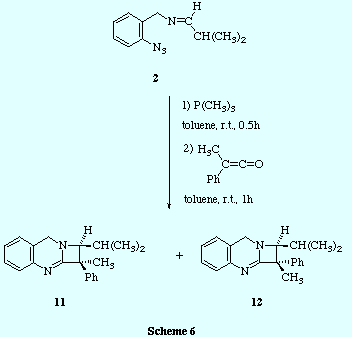
![]() Finally, the sequential treatment of imines 2 (those prepared by
reaction of o-azidobenzylamine 1 and aromatic aldehydes) with
trimethylphosphine and methylphenylketene led to the azetoquinazolines
10.
Finally, the sequential treatment of imines 2 (those prepared by
reaction of o-azidobenzylamine 1 and aromatic aldehydes) with
trimethylphosphine and methylphenylketene led to the azetoquinazolines
10.
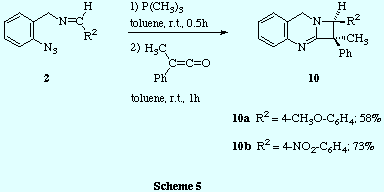
![]() 300 MHz 1H NMR spectra of crystallized compounds 10a and
10b revealed that, in both cases, only one diastereoisomer was
obtained. Similar analyses of the crude products also showed the same degree of
diastereoselectivity. The relative configuration between the two stereogenic
carbon atoms was ascertained by NOE difference experiments. Thus, irradiation
of the CH3-C1 signal induced enhancement of the H-C2 proton signal and
vice versa, thus showing that the methyl group at C1 and the hydrogen at C2 are cis.
300 MHz 1H NMR spectra of crystallized compounds 10a and
10b revealed that, in both cases, only one diastereoisomer was
obtained. Similar analyses of the crude products also showed the same degree of
diastereoselectivity. The relative configuration between the two stereogenic
carbon atoms was ascertained by NOE difference experiments. Thus, irradiation
of the CH3-C1 signal induced enhancement of the H-C2 proton signal and
vice versa, thus showing that the methyl group at C1 and the hydrogen at C2 are cis.
![]() The high diastereoselectivity observed in
the formation of the
azetoquinazolines 10 could be rationalized by
assuming that the reactive conformation of the imine-ketenimine is the one stabilized by pi-stacking
of the two aryl rings, that on the imine carbon atom and that on the carbon
atom of the ketenimine fragment.
The high diastereoselectivity observed in
the formation of the
azetoquinazolines 10 could be rationalized by
assuming that the reactive conformation of the imine-ketenimine is the one stabilized by pi-stacking
of the two aryl rings, that on the imine carbon atom and that on the carbon
atom of the ketenimine fragment.
![]() Experimental details.
Experimental details.
![]() References
References
1. Staudinger, H.; Hauser, E. Helv. Chim. Acta 1921, 4, 887
2. Krow, G. R. Angew. Chem. Int. Ed. Engl. 1971, 10, 435; Gambaryan, N. P. Russ. Chem. Rev. 1976, 45, 1251; Perst, H. Methoden Org. Chem. (Houben-Weyl) 4th ed., 1993, vol. E15, part 3, p. 2531-2710
3. Bernhard, A; Manfred, R. Angew. Chem. Int. Ed. Engl. 1979, 18, 320; Barbaro, G.; Battaglia, A.; Giorgianni, P. J. Org. Chem. 1987, 52, 3289
![]() Acknowledgements
Acknowledgements
We thank the Direccion General de Investigacion Cientifica y Tecnica (DGICYT) for financial support.