| [Molecules: 6] [Related articles/posters: 009 024 063 061 062 ] |
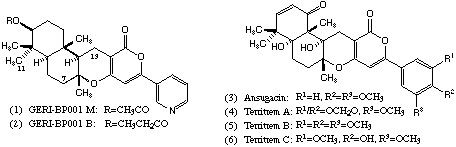
Because of the biological activities associated with these structures the development of synthetic routes that will allow rapid access to close analogues is of considerable interest and here we report some observations in this area which have enabled the synthesis of an analogue of the ACAT inhibitor GERI-BP001 M (1) in which the pyridine nitrogen has been replaced by carbon.
Of the two different synthetic approaches to these materials that have previously been reported[4,5], one involves construction of the tricyclic terpeniod portion (8) via a mercury(II) triflate-initiated polyene cyclisation, followed by subsequent introduction of the pyrone moiety[4] (Scheme 1, route A). We decided to examine whether a similar cyclisation process could be used to construct the entire tetracyclic core (10) in a single reaction pot (Scheme 1, route B).
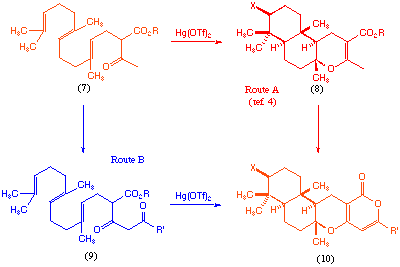
Scheme 1
The acyclic precursor for such a cyclisation would be compound (9), and as far as we are aware there are no examples of mercury(II) initiated cyclisation of such materials. Clearly there are a number of potential problems in using such a precursor, most notably the question of which of the three carbonyl functions will participate in the polyene cyclisation, and the question of how to encourage subsequent pyrone formation in the same reaction pot. Based on model studies we were reasonably certain that pyrone formation would take place if a tert-butyl ester function was used (9, R = But), we also anticipated that the desired keto-carbonyl should participate in the cyclisation under kinetically controlled conditions[6].
In order to investigate the cyclisation process we first required an effective route to the acyclic precursor (9). This was achieved in good overall yield as outlined in Scheme 2, and involved initial alkylation of tert-butyl acetoacetate (11) with farnesyl bromide, followed by C-acylation of the dianion derived from this product (12) with N-benzoyl-2-methyl aziridine[7]. We chose to introduce the benzoyl group since if the synthesis was successful, this would lead to an analogue of GERI-BP001 M in which the pyridyl substituent had been replaced by a phenyl group, and we were interested in the biological consequences of this change.
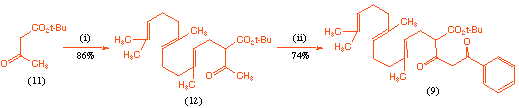
Scheme 2 Reagents and conditions: (i) NaH; farnesyl bromide, THF, room temp., 6 h; (ii) NaH; BuLi; N-benzoyl-2-methyl aziridine, 0 oC, 3 h
With the desired acyclic precursor (9) in hand we were able to study the to the mercury(II) initiated polyene cyclisation process. It was found that reaction of the acyclic precursor (9) with mercury(II) triflate[8] , followed by treatment with aqueous sodium chloride gave, after chromatography, the desired tetracycle (13) as the major product contaminated with variable quantities of what has tentatively been assigned as the tricycle (14). In addition to these products there were various less polar partially cyclised materials which could not be readily characterised.
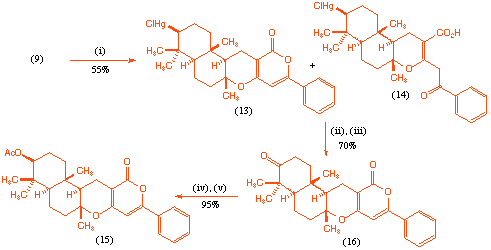
Scheme 3 Reagents and conditions: (i) Hg(OTf)2, DMA, CH3NO2, -20 oC, 2 h; NaCl, H2O, room temp., 48 h; (ii) NaBH4, O2, DMF, romm temp., 10 min; (iii) PCC, CH2Cl2, room temp., 30 min (iv) NaBH4, CH3OH, 0 oC, 15 min (v) Ac2O, pyridine, room temp., 16 h.
Although we were unable to further purify the tetracycle (13), treatment of the mixture with sodium borohydride in oxygen saturated DMF[9], followed by oxidation with PCC gave the ketone (16) which was readily isolated by chromatography in 70% overall yield. It appears that both (13) and (14) are converted into pyrone (16) via this sequence since no other products could be isolated from the oxidation process. Finally reduction of the keto-group with sodium borohydride, followed by acylation furnished the desired target structure (15) (Scheme 3).
The stereochemistry of product (15) was assigned on the basis of NOE experiments. Further evidence for this stereochemical assignment can be gained from comparison of characteristic signals in the 1H NMR spectrum (CDCl3) with those reported for the natural product GERI-BP001 M (pyripyropene E) (1). As can be seen, the terpenoid fragments of the two molecules give almost identical signals, which both supports the assigned stereochemistry, and emphasises the structural similarity of this region of the synthetic analogue.
