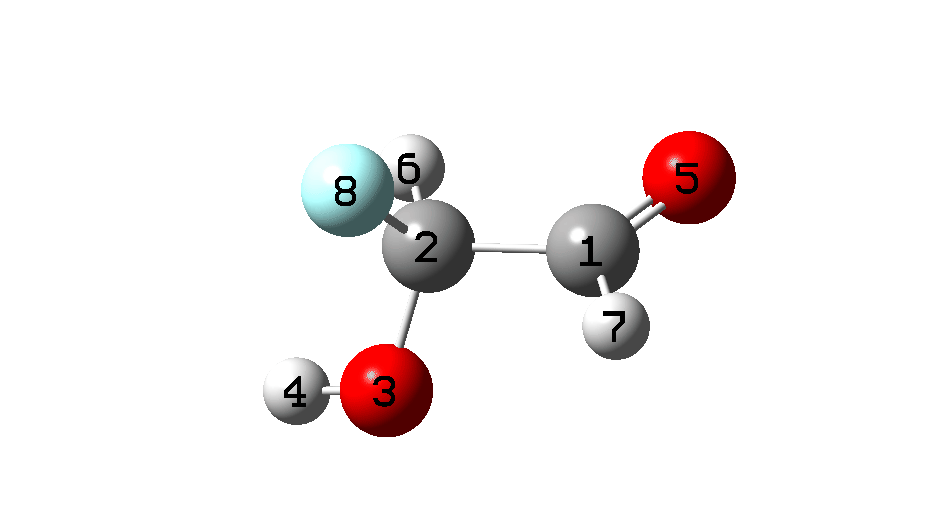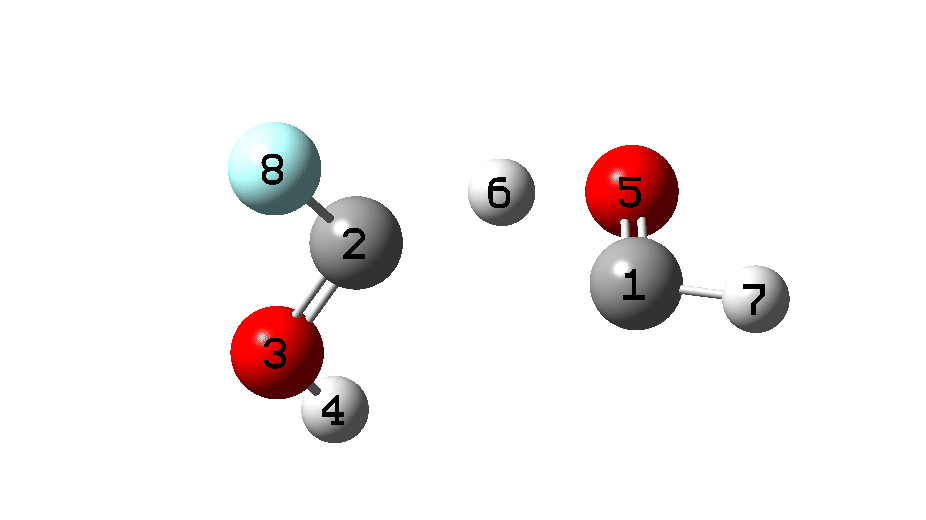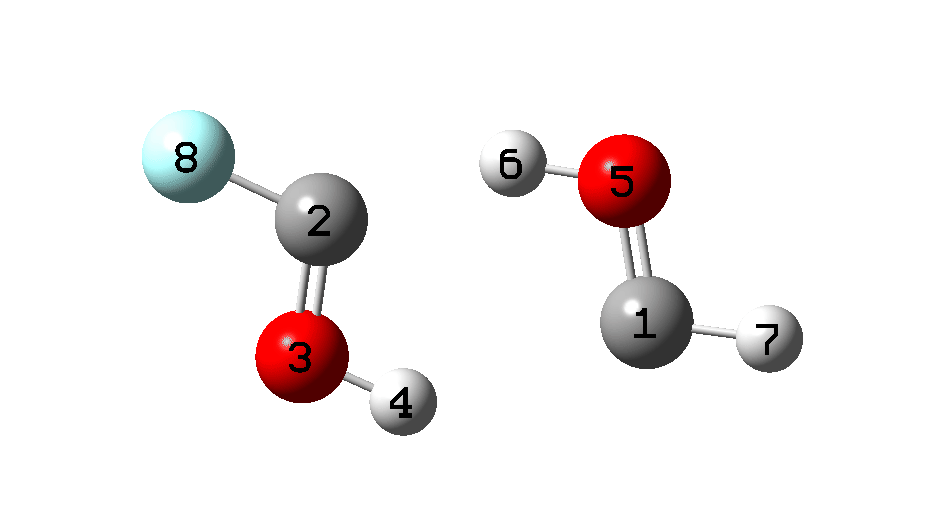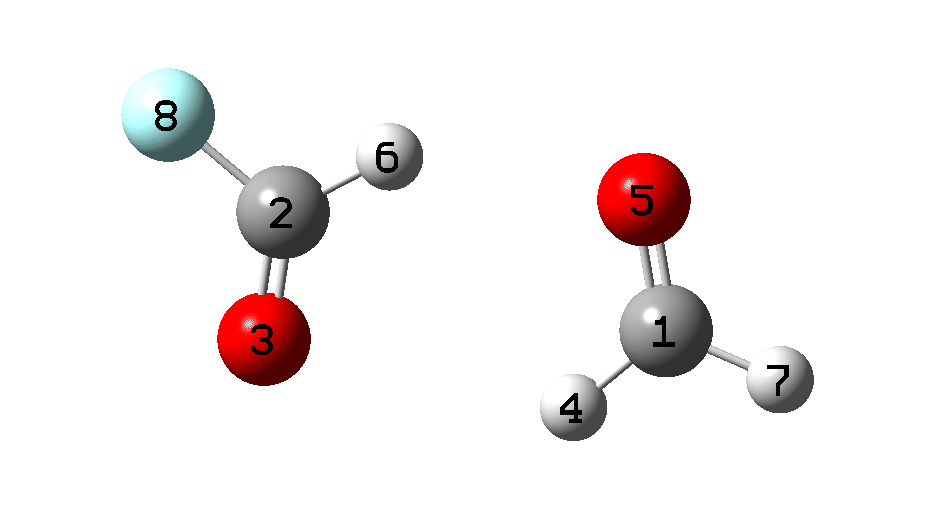
In 2008, the previously elusive hydroxycarbene, H-C-OH was finally reported[1] as having been captured by matrix isolation, accompanied by the observation that “we unexpectedly find that H–C–OH rearranges to formaldehyde with a half-life of only 2h at 11K by pure hydrogen tunnelling through a large energy barrier in excess of 30 kcal mol–1. A subsequent theoretical study of this tunnelling in 2017[2] reported that “the half-life calculation after monodeuteration is 2.97 × 1016 hours, which is extremely longer than before monodeuteration that is only 2.5 hours using the same calculation methods“; in other words a kinetic isotope effect kH/kD of ~1016, which is by far the largest ever suggested.[3] In 2011, the original study was extended to methylhydroxycarbene (X=Me)[4], again arguing for “Tunneling Control of a Chemical Reaction.” In this post,† I explore an alternative mechanism for rearrangement of hydroxycarbene to formaldehyde using a “double hydrogen transfer” via a dimeric transition state (Figure 1).

Figure 1. Two mechanistic possibilities for hydrogen transfer in hydroxycarbene.
There is general agreement that the rearrangement via a [1,2]-hydrogen shift (a four electron Woodward-Hoffmann “forbidden” pericyclic process) occurs with a barrier of > 30 kcal/mol. I will start with a traditional DFT method, ωB97XD/Def2-TZVPP/SCRF=dichloromethane) to see if I can replicate this assertion,[5],[6]‡ which yields 32.95 kcal/mol for the monomer free energy barrier. A CCSD(T)/Def2-TZVPP follow up gives 32.6 kcal/mol,[7] so we may presume that ωB97XD is a reasonable DFT method. These values represent a very slow thermal reaction. Kinetic isotope effects (using KINISOT, DOI: 10.5281/zenodo.10403662) for this reaction are listed below.
| KIE 1,2-H shift (ωB97XD) | KIE 1,2-H shift (CCSD(T) ) | Temp, K |
|---|---|---|
| 4.97 | 5.23 | 298.15 |
| 11.04 | 11.92 | 200.00 |
| 125.72 | 146.28 | 100.00 |
| 16,221.02 | 21,906.65 | 50.00 |
| 34,837,481,344.42 | 73,565,737,699.94 | 20.00 |
To locate a transition state for the dimer reaction, some subterfuge was used (for reasons that will become apparent). I needed a (computational) reaction that would generate two molecules of hydroxycarbene, which would then allow these two molecules to interact as they wished. Such a (hypothetical) reaction is shown in Figure 2.

Figure 2. Generation of two hydroxycarbene molecules from a precursor.
A transition state (X = Y = H, Figure 1) for this was located[8] which is -4.6 kcal/mol lower than the free energy of two molecules of hydroxycarbene at 298K and -13.7 kcal/mol lower at 20K.[9] At the CCSD(T)/Def2-TZVPP level @298K, the computed free energy of this TS[10] is -2.3 kcal/mol lower than two isolated monomers.
The located transition states are shown in Figure 3, and it consists of two hydroxycarbene molecules with a hydrogen bond formed between the hydrogen of one hydroxyl group and the carbene lone pair of the other hydroxycarbene.
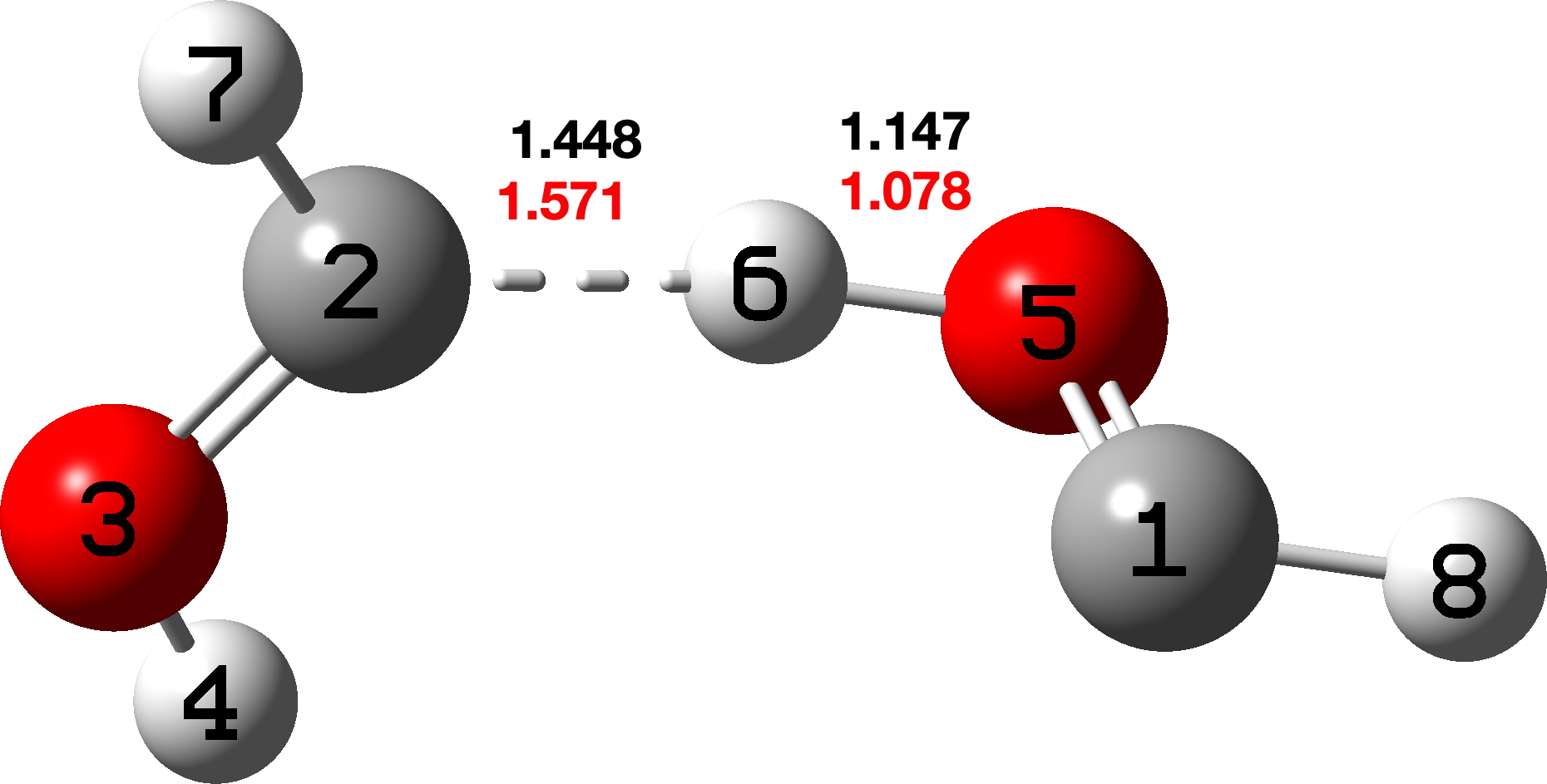
Figure 3. ωB97XD/Def2-TZVPP (red) and CCSD(T)/Def2-TZVPP (black) calculated TS for generation of two hydroxycarbene molecules. Click image for 3D model
There is support for such a hydrogen bond forming in the crystal structure database – see Figure 4.
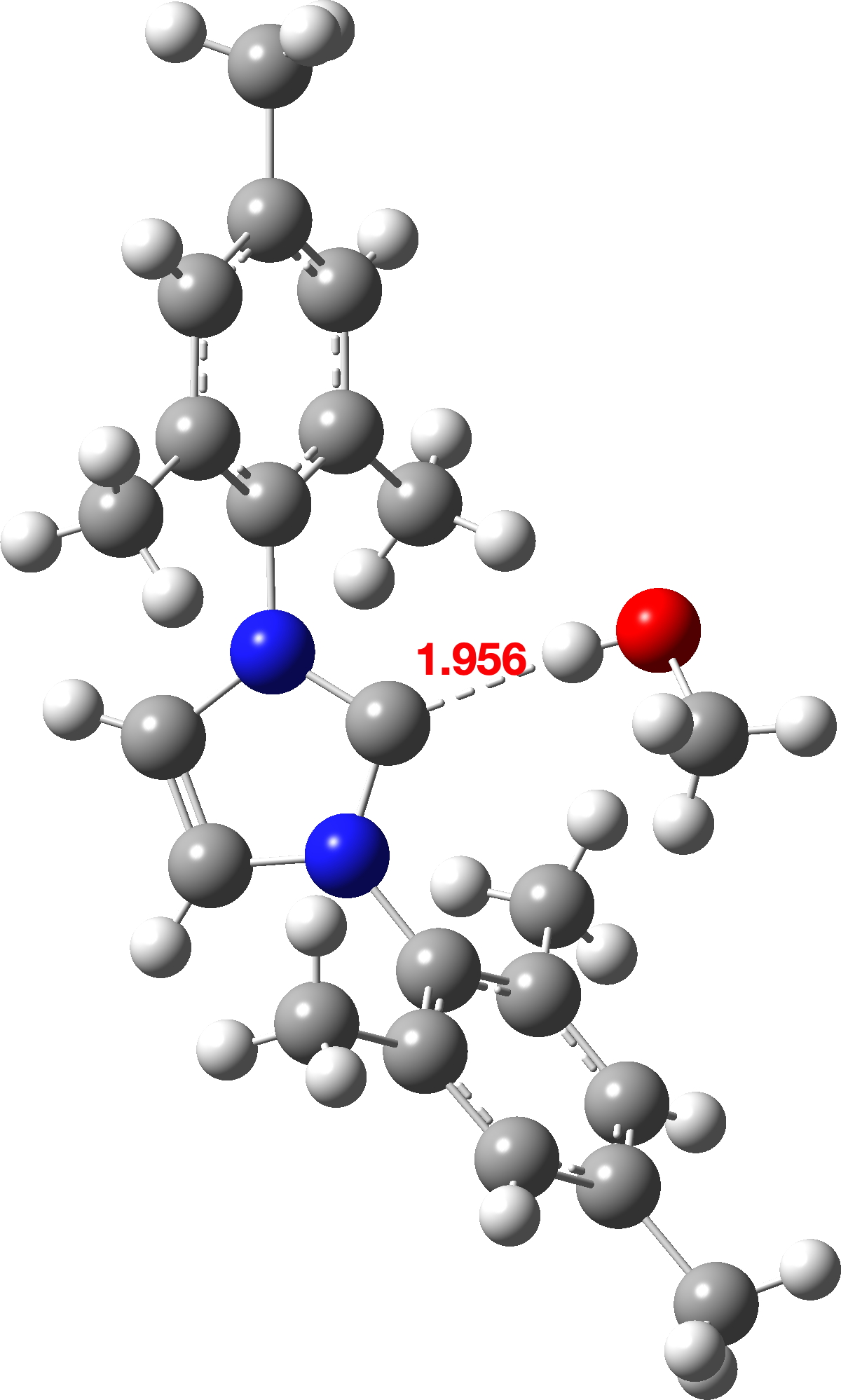
Figure 4. Crystal structure of an N-heterocyclic carbene with methanol.[11],[12]
An IRC (Figure 5) is needed to make more sense of the transition state. At this point, we need not concern ourselves about the preceding reaction profile (IRC 8 to 0), which as I mentioned was a computational subterfuge to generate two hydroxy carbene monomers in close proximity.

Figure 5. Intrinsic reaction coordinate for the reaction shown in Figure 2.[13]
It is what happens next that is crucial, which the IRC animation (Figure 6) makes clear. This is shown pausing at the TS and you should focus on what happens next, which is a rotation followed by two successive (but not entirely synchronous) proton transfers. As appropriate for a TS, the energy past this point only goes down.
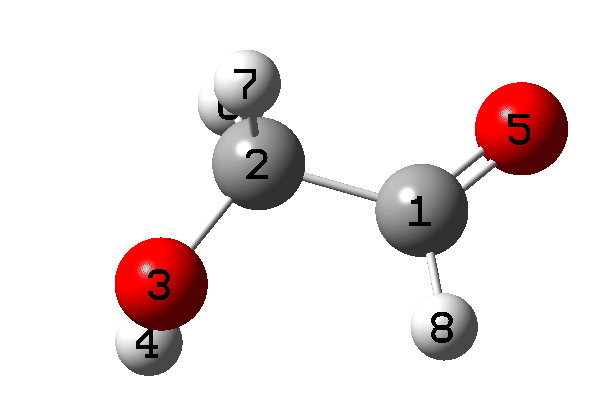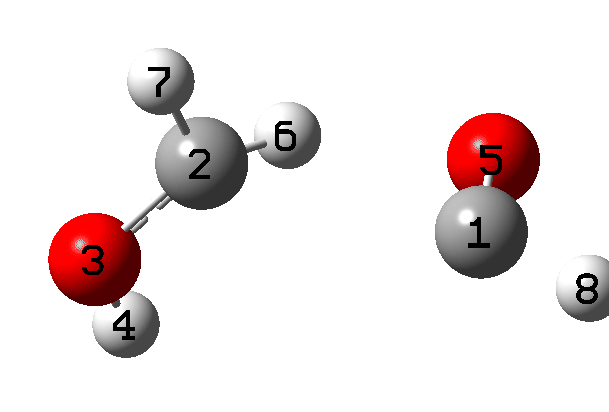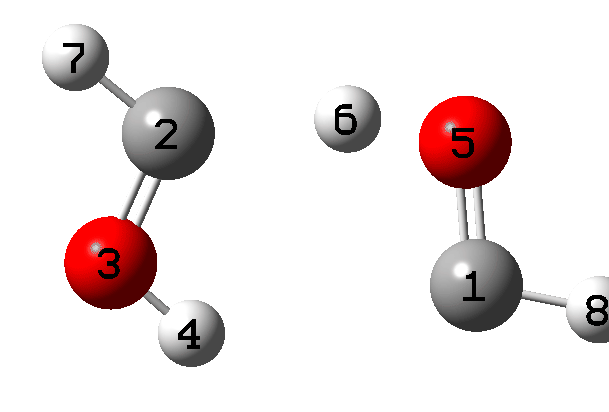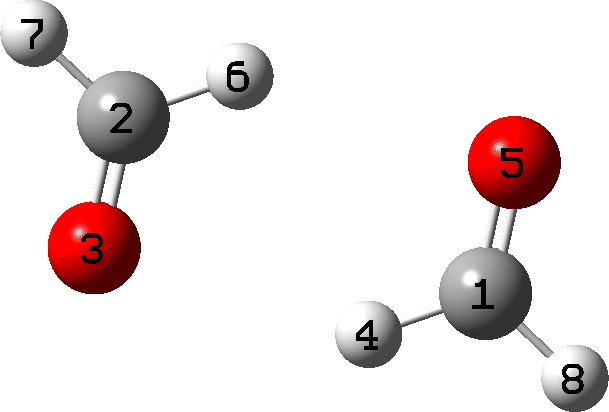
Figure 6. IRC animation ωB97XD/Def2-TZVPP for the reaction shown in Figure 2.[13]
Further insight can be found by inspecting the gradient norm of the IRC (Figure 7).

Figure 7. The calculated gradient normals along the IRC.
- From IRC 8 to 0, the generating reaction occurs (the “hidden intermediate” at IRC 1.5 is interesting but will not be discussed here).
- From IRC 0 to -4, a rotation of the two fragments occurs, setting up the hydrogen transfers.
- At IRC -5.5 the first hydrogen transfers.
- At IRC -6.1 the second hydrogen transfers.
The important observation is that at this stationary point (Figure 3), the computed free energy at 298K is -4.6 kcal/mol lower relative to two fully isolated hydroxycarbene molecules and it is even lower at 20K. We conclude from this analysis that when placed close to each other, two hydroxycarbenes react WITHOUT a barrier for exchanging hydrogens to form two molecules of formaldehyde. Hence the trick of generating the two hydroxycarbenes from a precursor to model this behaviour.
Kinetic isotope effects with deuterium substitution on both OH groups can be approximated using this new bimolecular transition state, via these outputs.[14],[8]
| HCOH: KIE (no tunnelling) | KIE (Bell tunneling) | Temperature, K |
|---|---|---|
| 3.366175 | 3.398699 | 298.15 |
| 5.622623 | 5.748042 | 200.00 |
| 25.480480 | 28.428388 | 100.00 |
| 504.392382 | – | 50.00 |
| 4,246,513.724875 | – | 20.00 |
I next look at a fluorinated version (X = Y = F).[15],[16]♥ The transition state[17] has C2h symmetry (Figure 8).
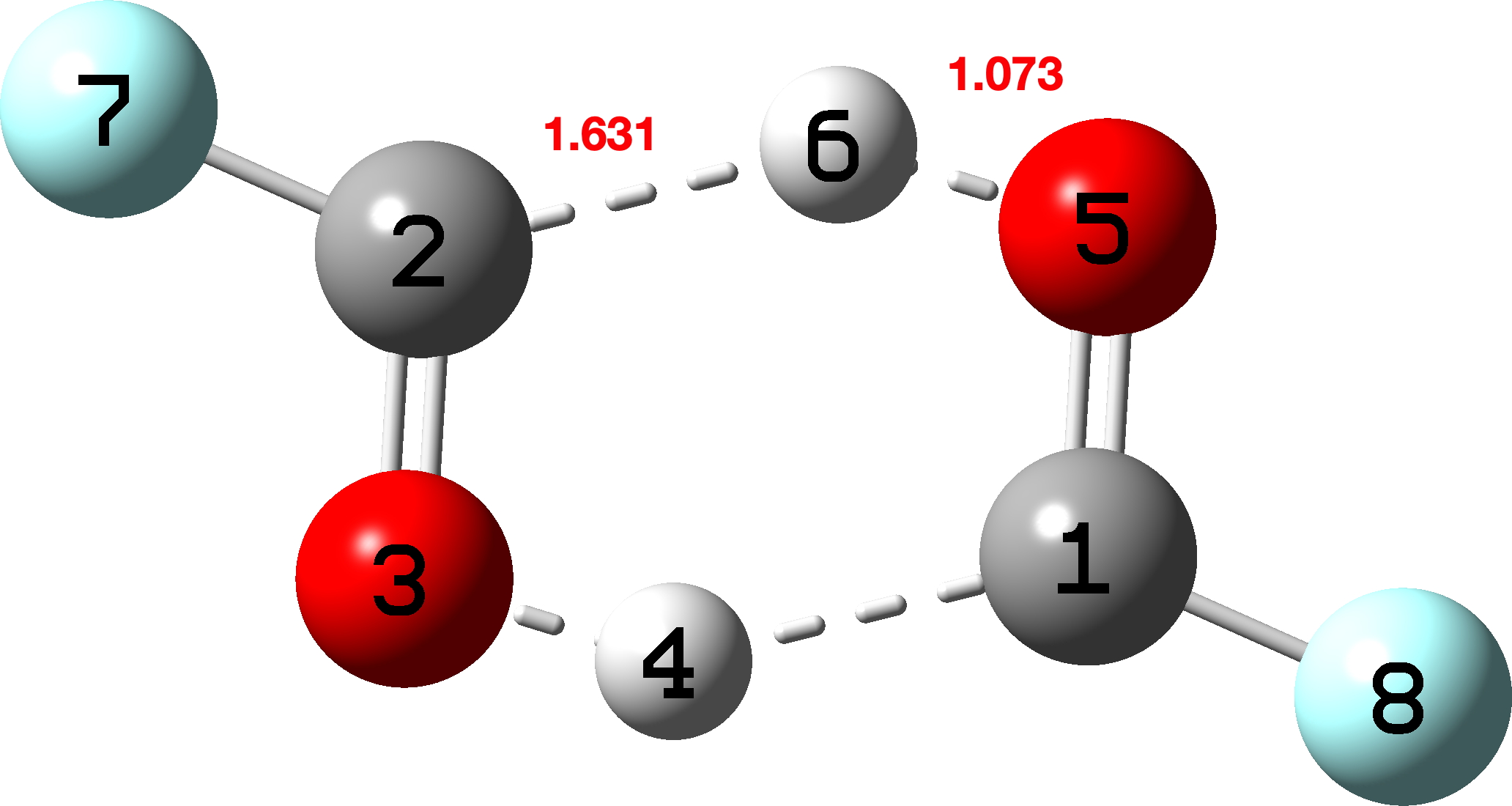
Figure 8. Transition state for synchronous double hydrogen transfer for X=Y=F.
The IRC[18] shows different behaviour (Figure 9, animation Figure 10). The dimer is a clear albeit very shallow intermediate now[15] – rather than just a point on the reaction coordinate as for X = Y = H, but the free energy of the TS (ωB97XD/Def2-TZVPP) is still lower by -4.4 kcal/mol compared to two isolated fluorohydroxycarbenes (ΔΔG -3.0 at the CCSD(T)/Def2-TZVPP level.[19]).

Figure 9. IRC for synchronous double hydrogen transfer for X=Y=F (Figure 1).
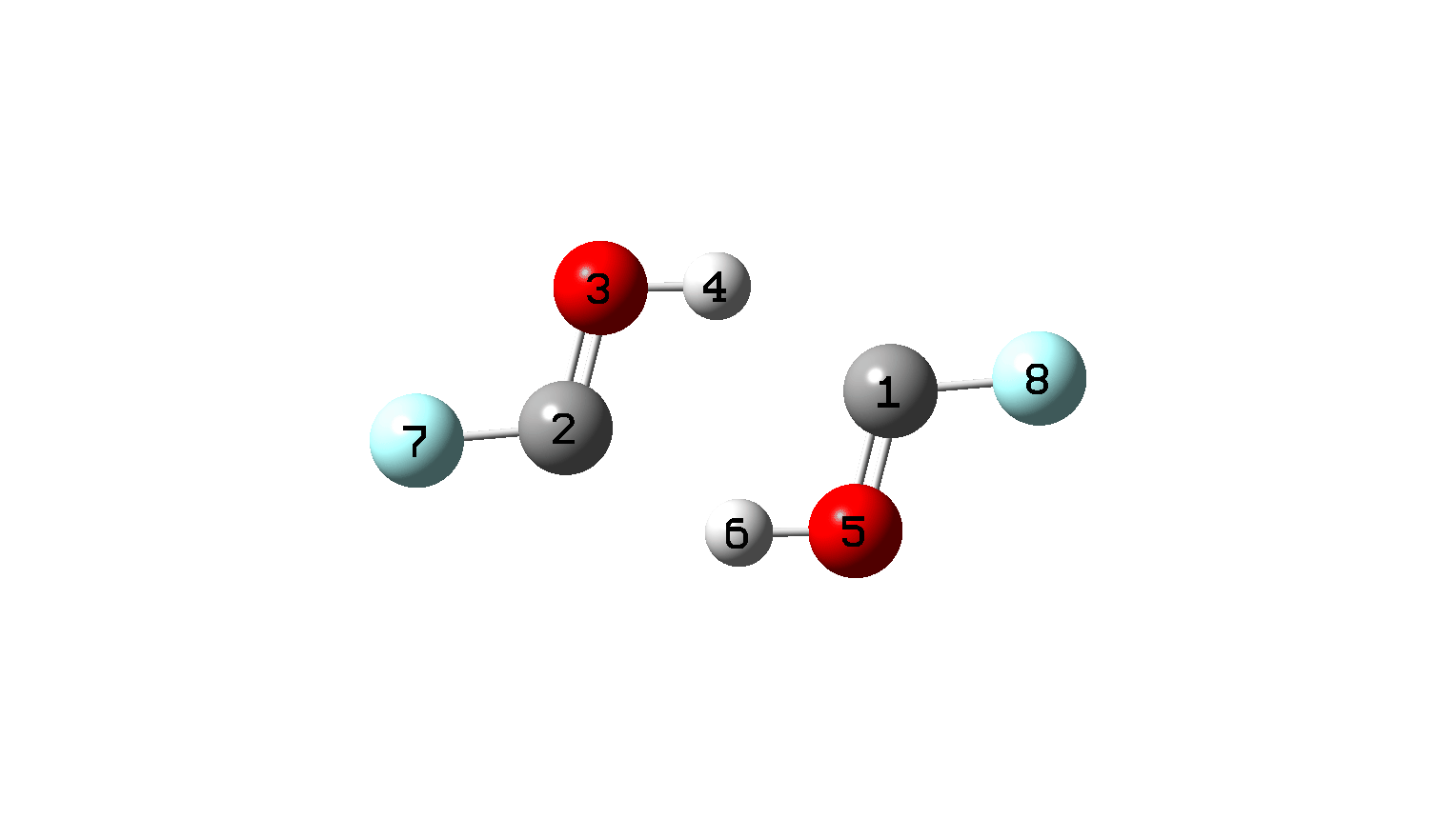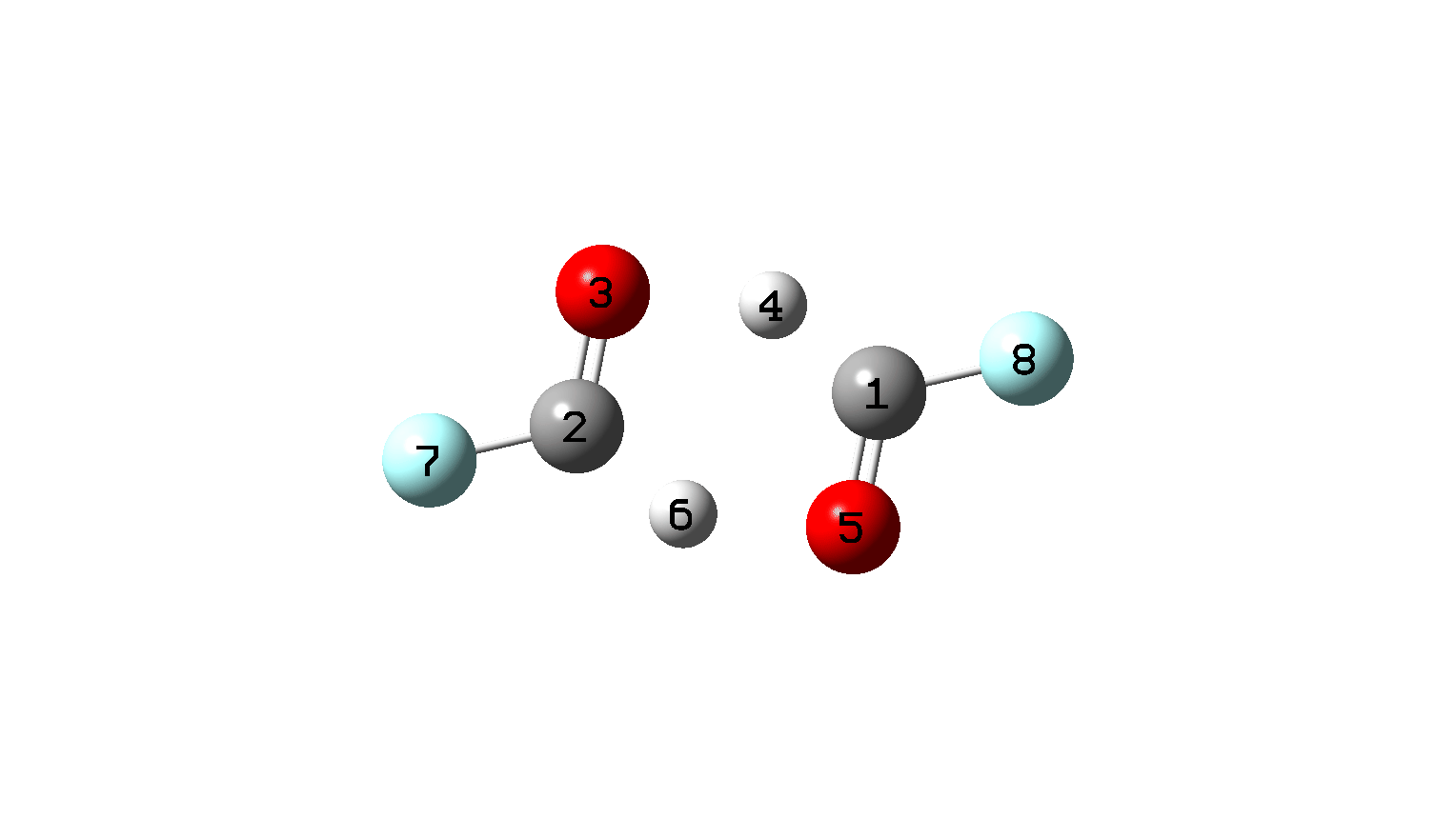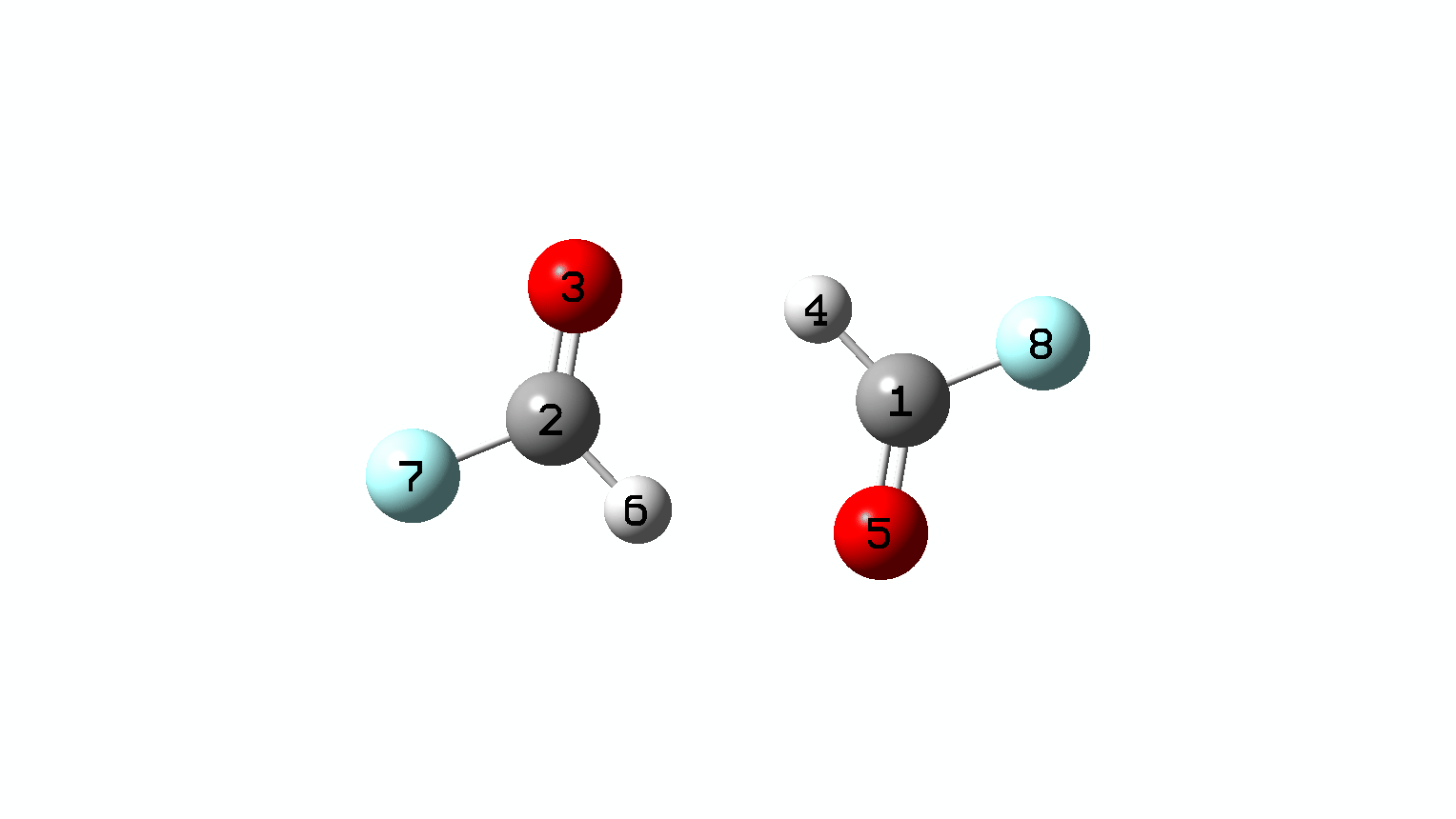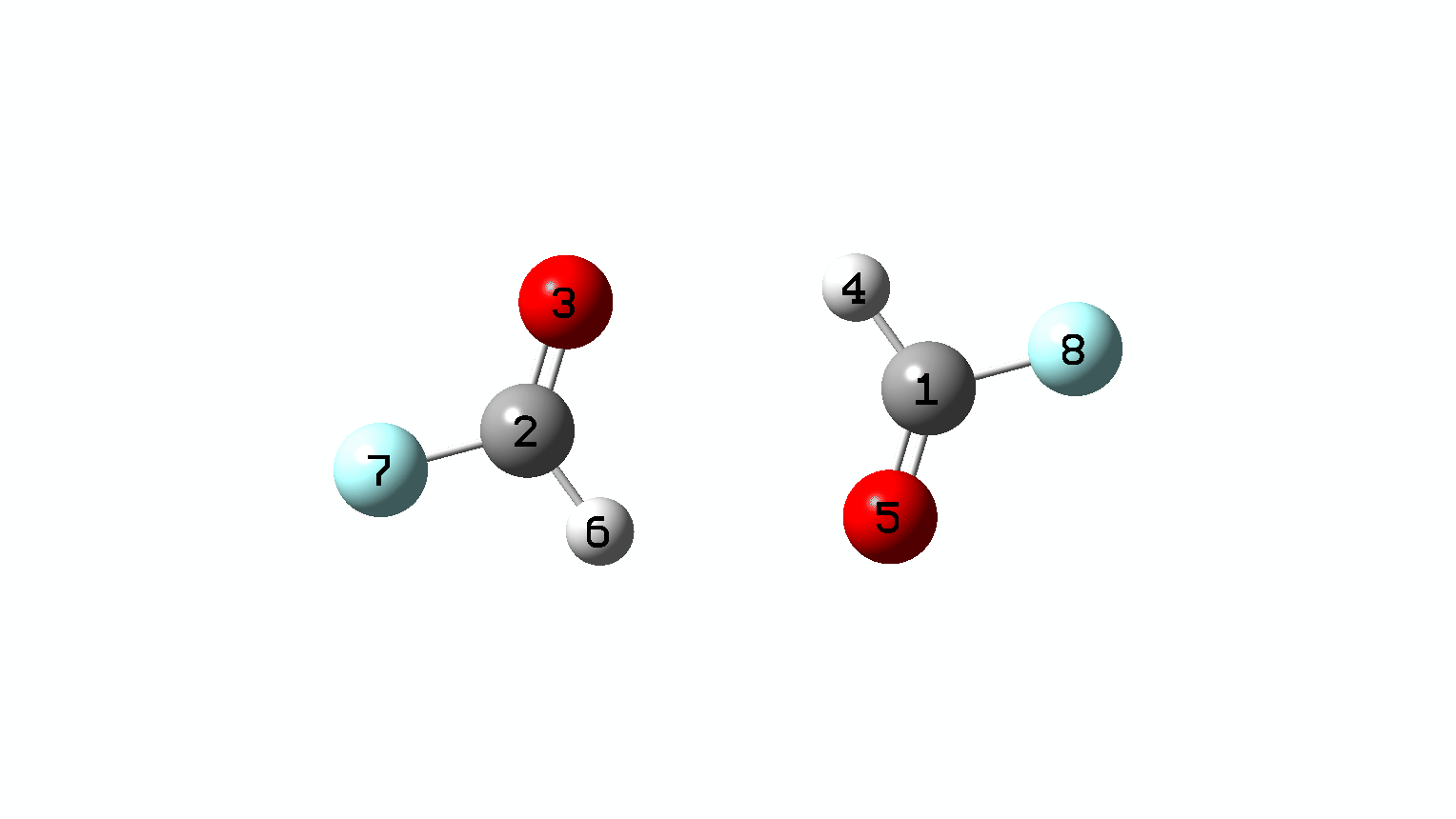
Figure 10. IRC animation for synchronous double hydrogen transfer for X=Y=F. (Figure 1)
The hydroxycarbene dimer itself is shown below (click image to view model)
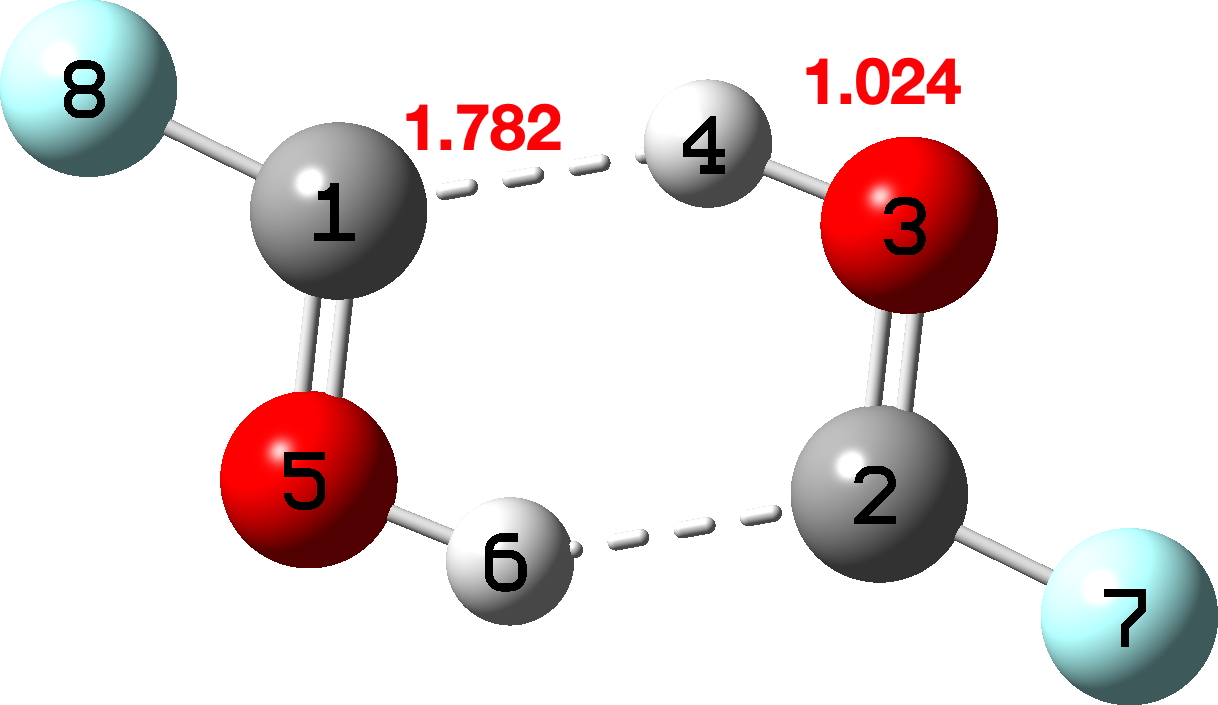
Figure 11. The structure of the FCOH H-bonded dimer.
The computed KIE are somewhat higher for the fluorinated molecule.[20],[9]
| FCOH: KIE (no tunnelling) | KIE (Bell tunneling) | Temperature, K |
|---|---|---|
| 6.587916 | 6.751828 | 298.15 |
| 14.724290 | 15.598102 | 200.00 |
| 187.689465 | 269.048707 | 100.00 |
| 31344.686809 | 32,350.352820 | 50.00 |
| 142,555,714,740.5 | – | 20.00 |
The conclusion is that whereas a unimolecular proton transfer to generate formaldehyde indeed passes (“tunnels”) through the significant barrier of a “forbidden” pericyclic reaction, an alternative bimolecular reaction is predicted to occur without a free energy barrier – the entropic penalty of combining two molecules is offset by the strong hydrogen bonds formed. Generating hydroxycarbene in a low temperature matrix suppresses the bimolecular mode, but when the matrix is warmed up, the two monomers can diffuse together to rapidly react. This speed can be achieved either through extreme tunnelling of one monomer, or by a barrierless concerted double hydrogen transfer via a dimer. Could it be that the fast disappearance of hydroxycarbene after formation might not be due to tunnelling control after all?
‡All the results are published as a FAIR data collection.[21] †This post has DOI:10.59350/syhqn-7md47[22] ♥A half-fluorinated reaction of HCOH + FCOH shows a similar profile to the non-fluoro version[23]

Author
References
- P.R. Schreiner, H.P. Reisenauer, F.C. Pickard IV, A.C. Simmonett, W.D. Allen, E. Mátyus, and A.G. Császár, "Capture of hydroxymethylene and its fast disappearance through tunnelling", Nature, vol. 453, pp. 906-909, 2008. https://doi.org/10.1038/nature07010
- N.D. Aisyah, R.N. Fadilla, H.K. Dipojono, and F. Rusydi, "A Theoretical Study of Monodeuteriation Effect on the Rearrangement of Trans-HCOH to H 2 CO via Quantum Tunneling with DFT and WKB Approximation", Procedia Engineering, vol. 170, pp. 119-123, 2017. https://doi.org/10.1016/j.proeng.2017.03.024
- H. Rzepa, "Reinvestigating the reported transition state structure of a concerted triple H-tunneling mechanism.", 2025. https://doi.org/10.59350/qgwfn-rsc92
- P.R. Schreiner, H.P. Reisenauer, D. Ley, D. Gerbig, C. Wu, and W.D. Allen, "Methylhydroxycarbene: Tunneling Control of a Chemical Reaction", Science, vol. 332, pp. 1300-1303, 2011. https://doi.org/10.1126/science.1203761
- H. Rzepa, "HCOH, Def2-TZVPP, G = -114.430626, 2G = -228.861252", 2025. https://doi.org/10.14469/hpc/15587
- H. Rzepa, "HCOH, 1.2-shift wB97XD/Def2-TZVPP, G = -114.378117 DG = 32.95", 2025. https://doi.org/10.14469/hpc/15596
- H. Rzepa, "HCOH, 1,2-shift CCSD(T)/Def2-TZVPP, G = -114.204230, DG = 32.6", 2026. https://doi.org/10.14469/hpc/15861
- H. Rzepa, "H-bonded hydroxycarbene dimer TS, wB97XD/Def2-TZVPP/SCRF=DCM, G = -228.868602 (1,2-TS + HCOH = -228.808743 , DG = 37.56 higher)", 2025. https://doi.org/10.14469/hpc/15586
- H. Rzepa, "H-bonded hydroxycarbene dimer TS, wB97XD/Def2-TZVPP/SCRF=DCM, G = -228.868602 @298 and -228.840293 @20K. DG = -13.68", 2026. https://doi.org/10.14469/hpc/15860
- H. Rzepa, "Hydroxycarbene TS, CCSD(T)/Def2-TZVPP, G = -228.516252 (2G monomer -228.512658, DDG = -2.26)", 2026. https://doi.org/10.14469/hpc/15874
- M. Movassaghi, and M.A. Schmidt, "N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols", Organic Letters, vol. 7, pp. 2453-2456, 2005. https://doi.org/10.1021/ol050773y
- Movassaghi, M.., and Schmidt, M.A.., "CCDC 274901: Experimental Crystal Structure Determination", 2005. https://doi.org/10.5517/cc971sb
- H. Rzepa, "H-bonded hydroxycarbene dimer TS, wB97XD/Def2-TZVPP/SCRF=DCM, G = -228.868548, IRC", 2025. https://doi.org/10.14469/hpc/15585
- H. Rzepa, "Two hydroxycarbene monomers 500A apart G = -228.863056 (G = -114.430626, 2G = -228.861252)", 2026. https://doi.org/10.14469/hpc/15856
- H. Rzepa, "H-bonded hydroxycarbene dimer, FCOH, wB97XD/Def2-TZVPP/SCRF=DCM, reactant dimer G = -427.484269", 2025. https://doi.org/10.14469/hpc/15593
- H. Rzepa, "H-bonded hydroxycarbene dimer, FCOH, wB97XD/Def2-TZVPP/SCRF=DCM, G = -427.486516", 2025. https://doi.org/10.14469/hpc/15590
- H. Rzepa, "H-bonded hydroxycarbene dimer, FCOH, wB97XD/Def2-TZVPP/SCRF=DCM, G = -427.486516, C2h symmetry G = -427.485832 DG = -3.95", 2025. https://doi.org/10.14469/hpc/15595
- H. Rzepa, "H-bonded hydroxycarbene dimer, FCOH, wB97XD/Def2-TZVPP/SCRF=DCM, IRC", 2025. https://doi.org/10.14469/hpc/15594
- H. Rzepa, "FCOH CCSD(T)/Def2-TZVPP dimer TS G = -426.898891, DG = -3.0 wrt two monomers", 2026. https://doi.org/10.14469/hpc/15929
- H. Rzepa, "FCOH, wB97XD/Def2-TZVPP/SCRF=DCM, IRC Separated reactant -427.489741( G = -213.739767, 2G =-427.479534)", 2026. https://doi.org/10.14469/hpc/15865
- H. Rzepa, "Hydroxycarbene", 2025. https://doi.org/10.14469/hpc/15584
- H. Rzepa, "The fast disappearance of hydroxycarbene through hydrogen tunnelling – or is it really tunnelling?", 2026. https://doi.org/10.59350/syhqn-7md47
- H. Rzepa, "FCOH + HCOH, TS G = -328.164440 IRC", 2025. https://doi.org/10.14469/hpc/15598
That’s a really interesting read, I hadn’t fully grasped the scale of the energy barrier involved. It makes you think about the limits of our observation methods.