A little while back, I wrote about anomeric-like effects in the sulfur ring S7.[1] I had started that exploration by retrieving the crystal structure from the ICSD (Inorganic crystal structure database) and then optimising these coordinates using a DFT method (MN15L/Def2-TZVPP to be precise). In demonstrating this effect to a student, I decided to create an initial guess for the molecule coordinates not from the crystal structure but by drawing and then minimising using a simple molecular mechanics force field – and only then subjecting it to DFT re-optimisation.[2] It turns out the result was quite surprising in one respect and so here I tell the rest of the story.
I start by noting that there is one fundamental difference between a DFT optimisation of the geometry and a molecular mechanics procedure – the latter cannot respond to (stereo)electronic orbital interactions, such as those found in anomeric effects. Thus DFT optimisation (using a simple opt keyword) starting from the mechanics coordinates leads – surprisingly perhaps – to a transition state rather than an equilibrium species. Normally the opt keyword does not produce such results – although to be certain of course the opt(calcfc) should be used to guarantee that a minimum rather than a transition state is found. The DFT optimised geometry has a C2 axis of symmetry, rather than the plane of symmetry expected for S7, running through atom 7 and the mid point of atoms 1 and 3 (Figure 1). All the S-S bond lengths are almost equal (Figure 1) – there is little discrimination and no anomeric effects are reflected in this geometry.
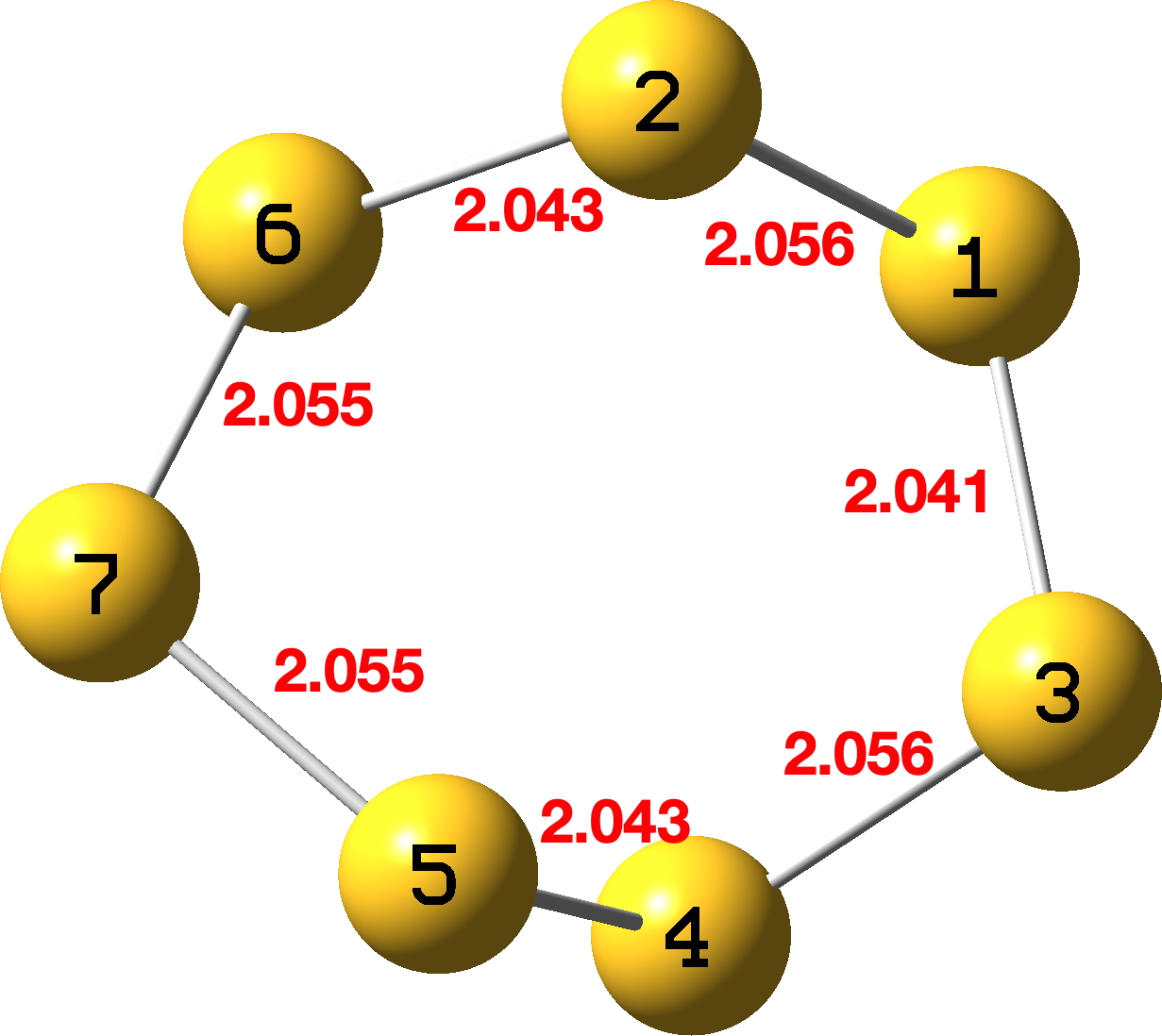
Figure 1.
To find out what the transition state connects, an IRC (intrinsic reaction coordinate calculation) was performed (Figure 2). It is symmetrical about the transition state, and leads to the known conformation of S7 in both directions, albeit with one strongly lengthened bond between S5-S7 on one side and between S6-S7 on the other side. As noted previously[1], this bond lengthening is a direct consequence of the anomeric orbital interactions. So the transition state is a low energy isomerisation, converting one anomeric isomer to the adjacent bond-lengthened one. To my knowledge, such a process has never been previously reported. It reminds one of mechanisms that exchange axial and equatorial positions in e.g. square planar or trigonal metal complexes.[3] (see also this link).

Figure 2.
The principle process occuring can be inferred by inspecting the dihedral angles S6-S7-S5-S4 and S2-S6-S7-S5 (Figures 3 and 4). The first changes from a dihedral close to 90° down to 0°, the second changes from 0° down to -90° and so directly relates to the orientation of a p-orbital on one sulfur and the adjacent S-S σ*-bond. The anomeric effect shifts by one bond during this process.

Figure 3.

Figure 4.
The process can be animated as in Figure 5.
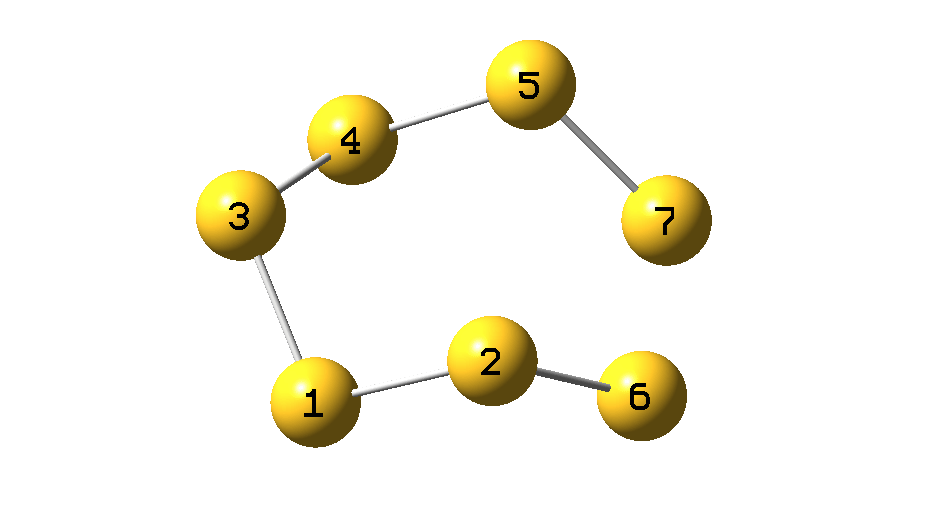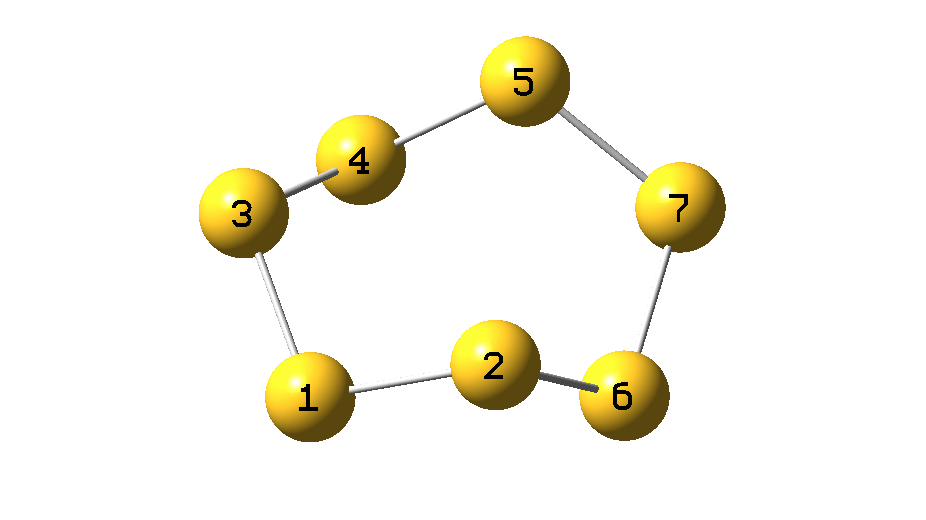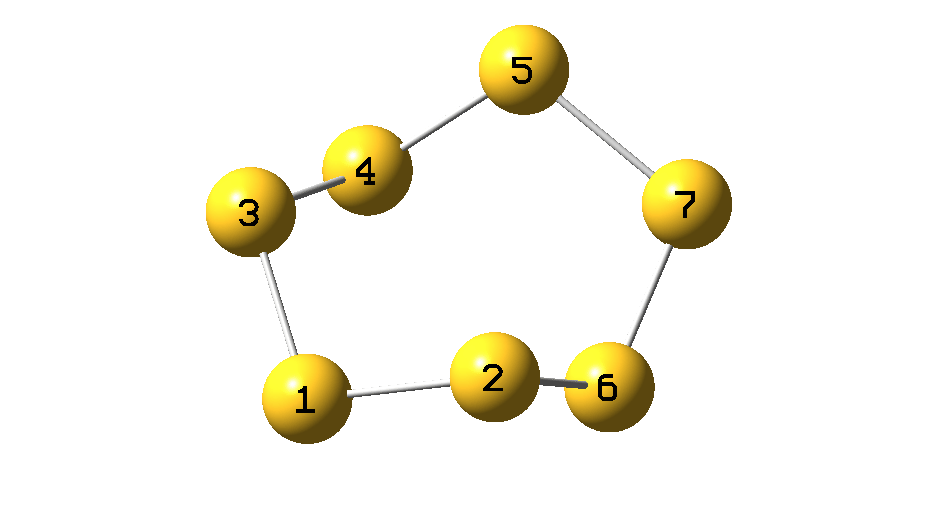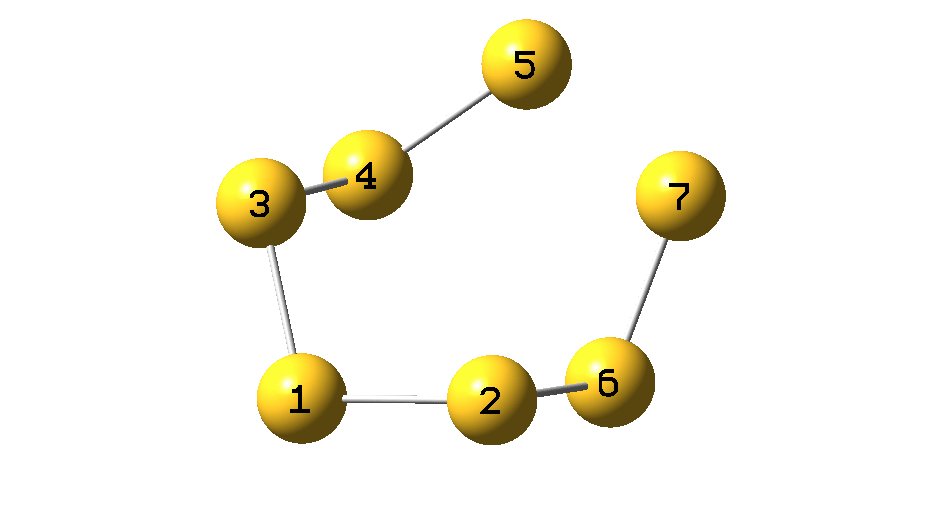
Figure 5.
The NBO7 orbital perturbation energies (kcal/mol) for transition state and equilibrium state[1] respectively are shown below. The former are all very close in value (note the absence of S7, through which the axis of symmetry passes) and hence induce no bond length discrimination, whereas the equilibrium state reveals the differences we have identified as an anomeric effect.
Transition state
LP S1 BD* S2-S6 5.81
LP S1 BD* S3-S4 7.17
LP S2 BD* S1-S3 6.82
LP S2 BD* S6-S7 7.33
LP S3 BD* S1-S2 7.17
LP S3 BD* S4-S5 5.81
LP S4 BD* S1-S3 6.82
LP S4 BD* S5-S7 7.33
LP S5 BD* S3-S4 5.53
LP S5 BD* S6-S7 5.27
LP S6 BD* S1-S2 5.53
LP S6 BD* S5-S7 5.27
Sum 75.9
Equilibrium geometry
LP S1 BD* S2-S6 5.03
LP S1 BD* S3-S4 7.08
LP S2 BD* S6-S7 12.34
LP S3 BD* S1-S2 7.06
LP S3 BD* S4-S5 7.04
LP S4 BD* S1-S3 7.08
LP S4 BD* S5-S7 5.03
LP S5 BD* S6-S7 12.34
LP S6 BD* S1-S2 10.09
LP S7 BD* S4-S5 10.09
Sum 83.2
This (accidentally discovered) transition state teaches us that the bond lengthening in S7 is directly associated with orbital orientations. And never to ignore a strange result – learning what happened can teach us a great deal.
Author
References
- H. Rzepa, "Cyclo-Heptasulfur, S<sub>7</sub> – a classic anomeric effect discovered during a pub lunch!", 2025. https://doi.org/10.59350/rzepa.28407
- H. Rzepa, "Anomeric isomerism in cyclo-heptasulfur.", 2026. https://doi.org/10.14469/hpc/15924
- H.S. Rzepa, and M.E. Cass, "A Computational Study of the Nondissociative Mechanisms that Interchange Apical and Equatorial Atoms in Square Pyramidal Molecules", Inorganic Chemistry, vol. 45, pp. 3958-3963, 2006. https://doi.org/10.1021/ic0519988