The recent report[1] of what is termed a “half-Möbius” molecule is generating a lot of excitement. It has its origins in a project to make odd-numbered cyclocarbons on STM (scanning tunnelling microscope) surfaces. I had discussed even-numbered cyclocarbons in another post[2], where I also happened to include several odd-numbered examples, such as C49 and C51. In this study[1] they were focussing on C13 and a precursor to this was to be C13Cl2. As part of the microscopy, they noticed this latter species was asymmetric (chiral) and so started the story of a “half-Möbius” molecule (molecules with twists in their topology are of course chiral). I should at this stage say that the concept of a half-Möbius is quite new and thought provoking. Perhaps the simplest way of explaining why, is that a conventional Möbius molecule (as with the strip or ribbon) requires two full circuits of the edge of the ribbon to return to the start, whereas this half version requires a full four circuits to achieve the same. More about this later.
Since STM microscopy is not capable of yielding accurate molecular geometries and bond lengths, the authors proceeded to calculate these – a non trivial undertaking! Basically, because of orbital degeneracies, the wavefunction has important multi-reference character. I thought I would illustrate some of the outcomes here. I actually start with a single reference calculation, using the r2scan-3c method,[3] which I had previously shown[2],[4] seemed to reliably reproduce geometries of even-numbered cyclocarbons such as C48, and also predicted the onset of bond length alternation (BLA) for rings of about 58 carbon atoms or greater. Applied to C13Cl2 r2scan-3c yields a planar molecule (Figure 1) with symmetrically disposed bond lengths.[5]
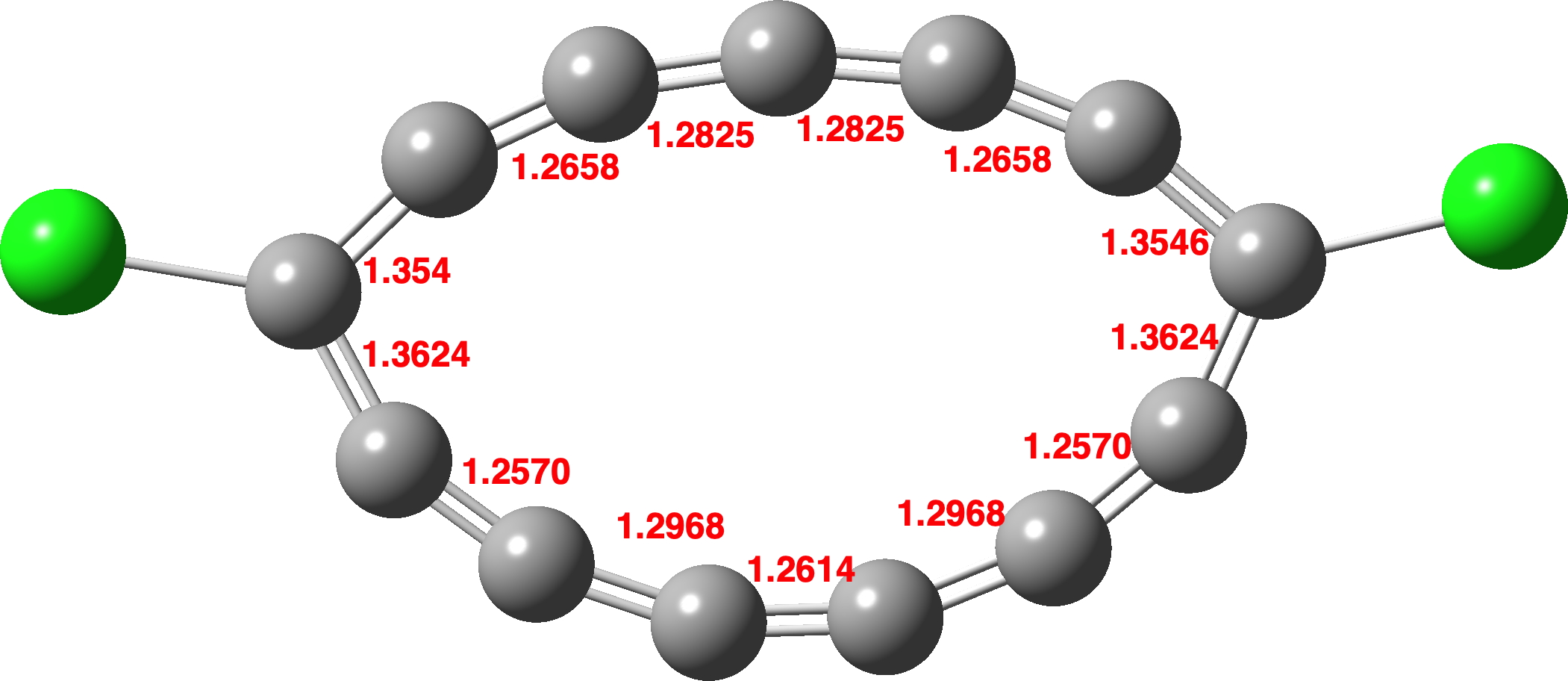
Figure 1. A r2SCAN-3c/Def2-mTZVPP optimised geometry for C13Cl2.[5]
The reported[1] calculation using CASPT2/cc-pVDZ[6] shows different behaviour (Figure 2). The singlet state geometry is asymmetric and non-planar, with interesting BLA in both the C6 and C7 fragments, but also more “aromatic-looking” values of ~1.40Å at the chlorine-connected carbon.

Figure 2. A CASPT2/cc-pVDZ gas phase optimised geometry for A: singlet C13Cl2 and b: Triplet.
A pure CASSCF(12,12) calculation performed here (Figure 3) using the Def2-TZVPP basis set (a triple-ζ basis – the geometry above is for a smaller double-ζ basis) reproduces both the non-planarity and the BLA, but not the aromatic-like bond lengths, confirming that a higher level of theory which includes MP2-like electron correlation perturbation corrections is needed for these deceptively simple molecules.
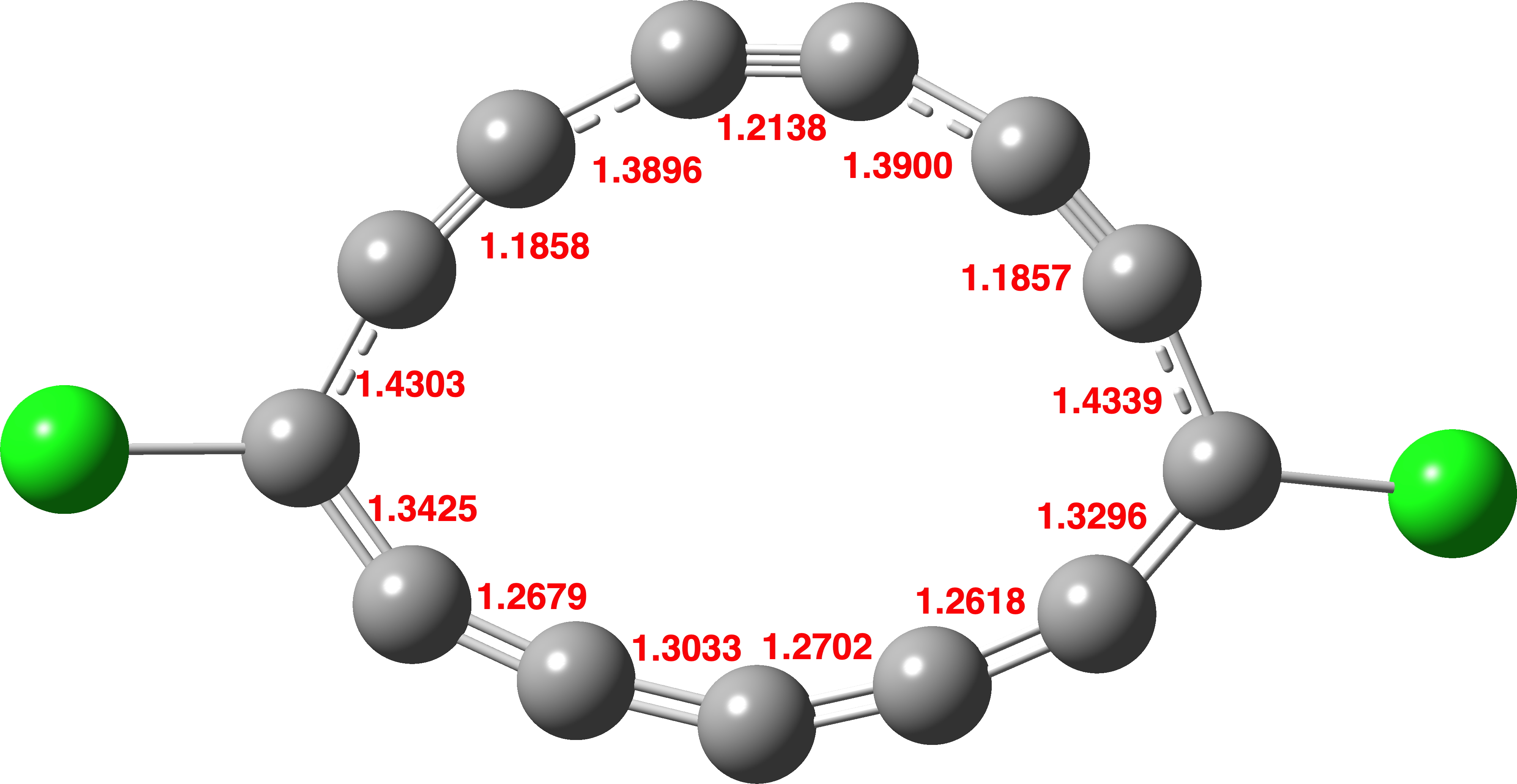
Figure 3. A CASSCF(12,12)/Def2-TZVPP calculation for singlet C13Cl2.
But what about that twist?
Now to the next stage of the story. Using orbitals derived from the wavefunctions, the authors[1] showed that only a 90° rotation (½π) occurred during a single trip around the edge of the molecular ribbon and hence 4*½π = 2π (360°) was required to achieve a return to the start. A full-Möbius molecule would achieve a 180° rotation or 1π for one circuit, and therefore requires only two circuits to achieve 2π. At this point, my thoughts turned to a well known topological theorem for these types of twisted systems[7], the Cãlugãreanu−White−Fuller theorem.[8]. This defines a topological invariant known as the linking number (Lk) which itself is the sum two quantities, the sum of local twists Tw and a writhe Wr. The latter can be thought of as the extent to which coiling of the central curve of the object can relieve local twisting of the ribbon. It is stated as:
Lk = Wr + Tw (each of which can be expressed in units of π).‡
One practical example is C14H14,[7], a molecule not entirely unrelated to C13Cl2. This has a “figure-eight”, lemniscular or “double-Möbius” topology (Figure 4).
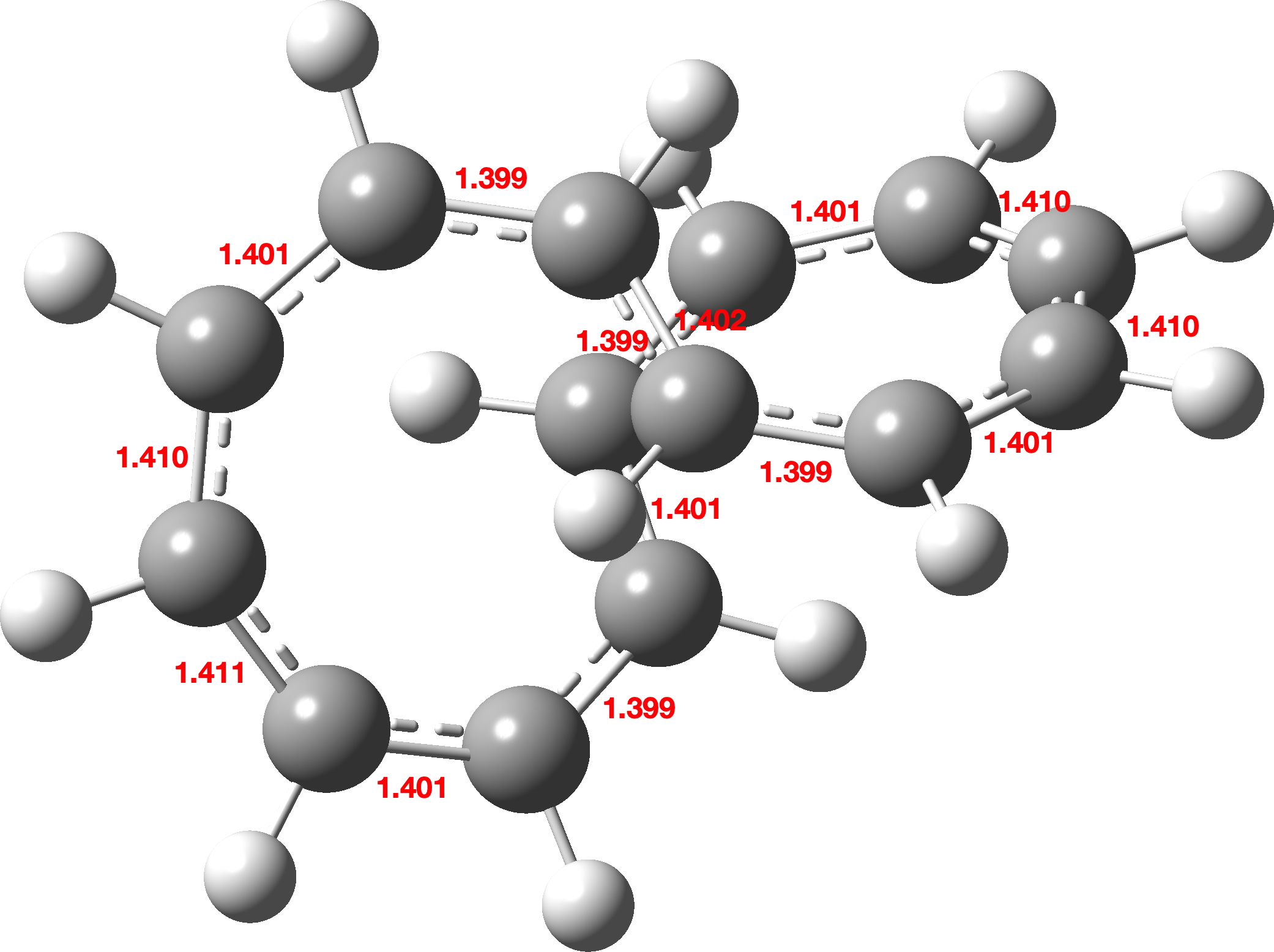
Figure 4: A double-twist Möbius annulene, calculated at the B3LYP/6-31G level. For the (almost identical) geometry using the more modern r2scan-3c/Def2-mTZVPP, see the FAIR data archive.[5]
When analysed using the expression above,[7] it shows values of Lk = 2π, Wr = 0.89π and Tw = 1.11π (B3LYP/6-31G* calculation).
Firstly, a bit of (unrecorded) history. When I discovered this little lemniscular lovely, I did what had been done above, namely I added the total rotation of the orbital basis (the p-π-orbitals on each carbon) and returned to my starting point in just one circuit. I obtained a rotational sum of ~180°, or ~π. I knew that returning to the starting point in one circuit (2π) meant it was not a conventional Möbius molecule but a “figure-eight” or double twist topological isomer (which Möbius, along with Listing, had identified!). This form had, not C2 symmetry as per a single twist Möbius, but the higher D2 chiral symmetry. The correct answer for this twist sum was therefore surely 2π, not 1π? I had lost ~1π worth of twist! And this is when I came across the above theorem, which put simply indeed allows any fraction of twist to be “lost” by its conversion into writhe, as can be seen from the values shown above.†
So here is my question. Might it be possible that the same has happened to C13Cl2? A measured orbital rotation of ~90° or ~½π (and hence the term half-Möbius) would only be correct if the writhe for this molecule – the coiling of the central curve out of a plane – was zero. If instead the writhe also had a value of lets say ~½π, then
Lk = ~1π, comprising Wr = ~½π and Tw = ~½π
which would make it a conventional rather than half-Möbius molecule.
To conclude: the reported interpretation of C13Cl2 as a “half-Möbius” molecule is only correct if it does not “writhe” topologically to any significant extent. Watch this space for updates!
‡Lk can be both a positive or a negative integer, depending on which enantiomer is used, and hence acts as a chiral descriptor in the manner of the Cahn-Ingold-Prelog convention. †Wr and Tw do not have to have the same sign. Thus the value of Tw can be greater than that of Lk if Wr is opposite in sign. For an extreme example of these various effects see here.[9]. The proposed molecular trefoil knot has values of Lk 6π = Tw -0.8π + Wr +6.8π Not only are the twist and writhe of opposite sign, the knot is composed almost entirely of writhe and no twist!
This post has DOI: 10.59350/5q3ka-2ag71
Author
References
- I. Rončević, F. Paschke, Y. Gao, L. Lieske, L.A. Gödde, S. Barison, S. Piccinelli, A. Baiardi, I. Tavernelli, J. Repp, F. Albrecht, H.L. Anderson, and L. Gross, "A molecule with half-Möbius topology", Science, vol. 392, 2026. https://doi.org/10.1126/science.aea3321
- H. Rzepa, "Molecules of the year 2025: Cyclo[48]carbon and others – the onset of bond alternation and the Raman Activity Spectrum.", 2025. https://doi.org/10.59350/g4309-gv109
- S. Grimme, A. Hansen, S. Ehlert, and J. Mewes, "r2SCAN-3c: A “Swiss army knife” composite electronic-structure method", The Journal of Chemical Physics, vol. 154, 2021. https://doi.org/10.1063/5.0040021
- H. Rzepa, "Quantum crystallography: The structure and C-C bond length alternation of [18]-annulene.", 2026. https://doi.org/10.59350/k0kjg-hpc66
- H. Rzepa, "The first "half-Möbius" molecule: A question about its twist?", 2026. https://doi.org/10.14469/hpc/15786
- I. Rončević, A. Baiardi, and F. Paschke, "Supplementary data for A Molecule with Half-Twisted Möbius Topology", 2025. https://doi.org/10.5281/zenodo.15495263
- S.M. Rappaport, and H.S. Rzepa, "Intrinsically Chiral Aromaticity. Rules Incorporating Linking Number, Twist, and Writhe for Higher-Twist Möbius Annulenes", Journal of the American Chemical Society, vol. 130, pp. 7613-7619, 2008. https://doi.org/10.1021/ja710438j
- G. Călugăreanu, "On isotopy classes of three dimmensional knots and their invariants", Czechoslovak Mathematical Journal, vol. 11, pp. 588-625, 1961. https://doi.org/10.21136/cmj.1961.100486
- H. Rzepa, "Chemistry with a super-twist: A molecular trefoil knot, part 1.", 2010. https://doi.org/10.59350/j60gh-gzr35