Recently, the 100th anniversary of the birth of the famous chemist Derek Barton was celebrated with a symposium. One of the many wonderful talks presented was by Tobias Ritter and entitled “Late-stage fluorination for PET imaging” and this resonated for me. The challenge is how to produce C-F bonds under mild conditions quickly so that 18F-labelled substrates can be injected for the PET imaging. Ritter has several recent articles on this theme which you should read.[1],[2].
The resonance was that back in 1999 I had been collaborating with colleagues to study the mechanism of rapid fluoridation using R2IF reagents.[3]. This article, by the way, contains a very early example of the use of FAIR data (see here).‡ A further resonance is that Ritter computes that the displacement of an aryl-O bond by a nucleophilic fluorine is a concerted process, unlike the stepwise Meisenheimer like complexes normally occurring in nucleophilic aromatic substitutions. A few years back I explored the possibility of concerted nucleophilic substitutions, finding that F in particular was very prone to such behaviour. So it is nice to see Ritter’s real-world example of such a mechanism and indeed that his reagents (PhenoFluor) represent a significant improvement on the R2IF ones we had been exploring.

To celebrate this new chemistry, I include some results of my own which augment Ritter’s. Firstly I should start with the structure of the reagent, which contains a carbon surrounded by four heteroatoms. There are few such motifs known.♥ Thus a carbon attached to two N, one F and one O has no reported crystal structures. Relaxing the criterion to two N, one F and one other offers 71 examples, of which the most interesting are the outliers with C-F distances > 1.4Å.
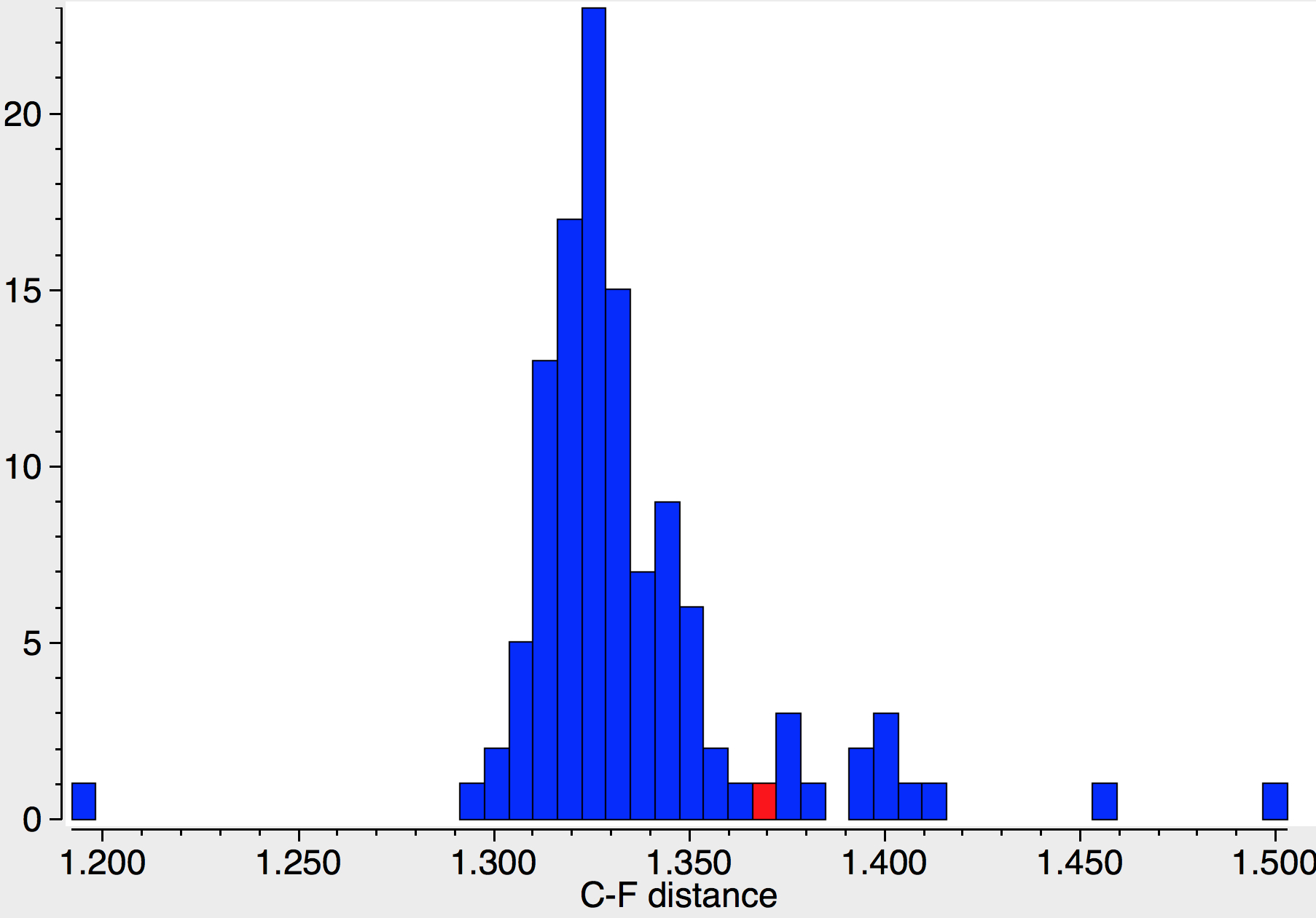
The one with the value 1.5Å (DOI: 10.5517/ccdc.csd.cc1njjg5) is probably an error, as is the one at 1.45Å[4]. So it was a surprise to find that the calculated structure of PhenoFluor (R=2,6-di-isopropyl, B3LYP+D3BJ/Def2-TZVPPD/SCRF=toluene) had a C-F distance of 1.456Å which is surely a candidate for the longest known C(sp3)-F bond.† The computed Wiberg C-F bond order is 0.687, which is well reduced from a single bond order. This is probably due to strong anomeric effects from both nitrogens and the one oxygen, which “gang up” on the fluorine to weaken its bond and expel it as a nascent fluoride anion. Thus the E(2) NBO interaction energy is 25.1 kcal/mol for the N(Lp)-CFσ* interaction, which is unusually large, whereas the N(Lp)-COσ* interaction is only 9.2 kcal/mol.
The fluoridation is indeed computed as a concerted process, the IRC animation being shown below. Note that the trajectory of the F is initially away from the carbon but not towards the aryl group. Here it is simply forming a “hidden” fluoride anion intermediate. The trajectory then changes direction to attack the ipso-carbon. So it is a concerted but two-stage reaction path.
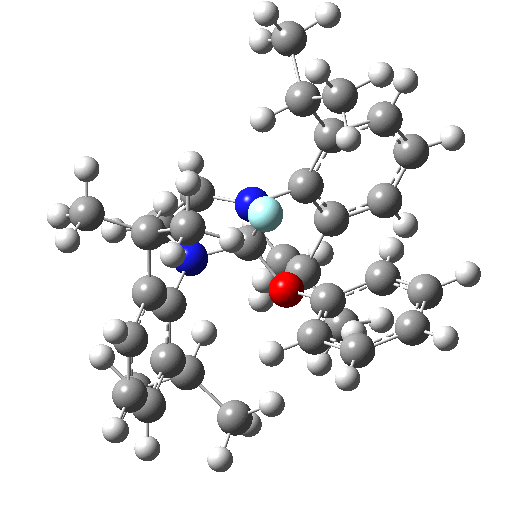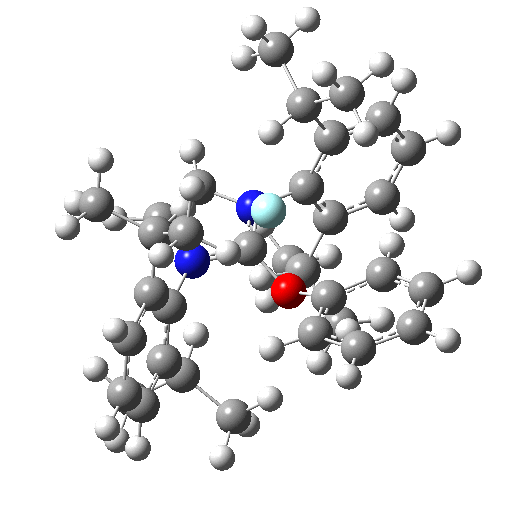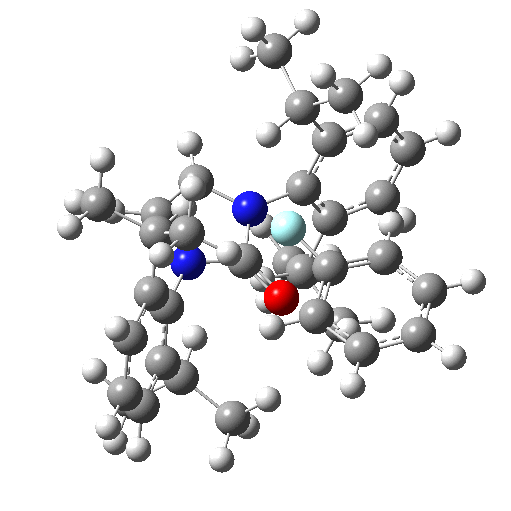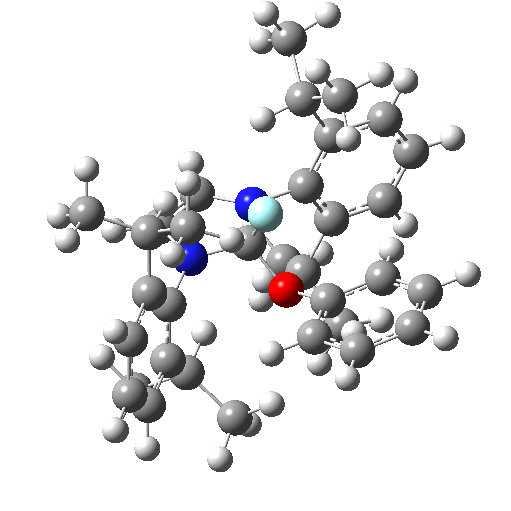
The IRC energy profile corresponds to a free energy barrier of 23 kcal/mol, as reported by Ritter.

Here is a less well used property along the reaction path, the dipole moment response. This shows a very abrupt charge separation in the region of the transition state and its collapse shortly after which suggests that the reaction barrier might be sensitive to the polarity of the solvent.

The issue then arises as to how much aromatic resonance is lost at the transition state. A NICS(1) aromaticity probe placed 1Å above the centroid of the aryl ring has the value -9.3 ppm, close to the value of ~ -10ppm for benzene itself. So this relatively facile reaction is in part due to significant preservation of the aryl stabilisations by aromatic resonance.
To close, the Phenofluor reagent is commercially available as a 0.1M solution in toluene, which makes one wonder if it is possible to obtain crystals. It might be of course that when the solution is concentrated, it reverts to the iminium fluoride ion pair shown above. But if crystals are possible, then it would be interesting to verify that the C-F bond in this species is indeed unusually long, perhaps even a record holder?
‡The data represent an early use of the Chime plugin to present a visual 3D model. I really should re-work that page to allow use of eg JSmol, enabled here on this blog.
♥Excepting CF3X motifs.
†FAIR data for the results reported here can be found at DOI: 10.14469/hpc/4713
Author
References
- P. Tang, W. Wang, and T. Ritter, "Deoxyfluorination of Phenols", Journal of the American Chemical Society, vol. 133, pp. 11482-11484, 2011. https://doi.org/10.1021/ja2048072
- C.N. Neumann, and T. Ritter, "Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent", Accounts of Chemical Research, vol. 50, pp. 2822-2833, 2017. https://doi.org/10.1021/acs.accounts.7b00413
- M.A. Carroll, S. Martín-Santamaría, V.W. Pike, H.S. Rzepa, and D.A. Widdowson, "An ab initio and MNDO-d SCF-MO computational study of stereoelectronic control in extrusion reactions of R2I–F iodine(III) intermediates†", Journal of the Chemical Society, Perkin Transactions 2, pp. 2707-2714, 1999. https://doi.org/10.1039/a906212b
- T.N. Bhat, and H.L. Ammon, "Structure of N,N,N'N'-tetrakis(2-fluoro-2,2-dinitroethyl)oxamide by the consistent electron density approach", Acta Crystallographica Section C Crystal Structure Communications, vol. 46, pp. 112-116, 1990. https://doi.org/10.1107/s0108270189005044
Tags: Antimatter, Barton, Chemistry, Derek Barton, Fluorine, Medical physics, Medicinal radiochemistry, Natural sciences, Neuroimaging, Physical sciences, Positron emission tomography, Radiation therapy, Ritter, Tobias Ritter
Tobias Ritter has alerted me to another recent article by Eric Jacobsen entitled Concerted nucleophilic aromatic substitutions, where carbon kinetic isotope effects are used to determine the characteristics of the rate determining step (DOI:10.1038/s41557-018-0079-7) Those involving fluorine as nucleophile tend to be concerted!
It’s not just SNAr reactions with fluorine that are concerted–it’s all SNAr reactions. In fact, those with fluorine are more likely to be stepwise, which makes Tobias’ example all the more remarkable.
Eugene
Could you provide any literature citations for that assertion? As Tobias alerted me to, Eric Jacobsen (see previous comment) starts with the statement “The generally accepted mechanism for SNAr reactions involves a two-step addition–elimination sequence via a discrete, non-aromatic Meisenheimer complex” but he goes on to demonstrate three classes of mechanism, A, B and C, spanning truly stepwise, concerted and borderline. So the claim that ALL SNAr reactions are concerted is perhaps too strong.
Ah, forgive me–I was being hyperbolic. What we showed in the study you mentioned is that most SNAr reactions are concerted. To have a stepwise mechanism, you need to have both an electron-deficient ring (to stabilize the charge) and a poor leaving group (and fluoride is terrible). So I should have said: it’s not just SNAr reactions with fluorine that _tend to be_ concerted, it’s all SNAr reactions.
By the way, I should mention that your blog post from 2013 on this subject was one of the things that got me thinking about this topic. So thank you!
Eugene, forgive me for not connecting you as a co-author on DOI: 10.1038/s41557-018-0079-7!
By the way, I have made some initial attempts at correlating the NBO (E2) perturbation energy with the ease of delivery of a fluoride anion. The value of 25 kcal/mol might be a good metric for optimising the substituent on each nitrogen. I did try replacing the 2,6-DIP group with TMS (recognising that F has a big affinity for Si!). I wonder how many different N substituents were investigated by Ritter and co. before they settled on 2,6-DIP. It would be good to know what all the rejects were!
I would hazard a guess that the 2,6-diisopropylphenyl was probably one of the first they tried! After all, IPr and IMes are probably the most commonly used NHCs, so everything needed for the synthesis was probably close at hand.
By the way, the observation that concerted SNAr reactions are common suggests that hyperconjugation of the two transition state bonds forming an breaking in concert with the ring must be especially favourable, so as to lead to relatively little loss of aromaticity. Thus facile concerted SNAr reactions may simply be related to a high degree of retention of aromaticity.
We thought so too, but when analyzed the minimum energy path for our concerted reaction, we found that the NICS parameters indicated the loss of aromaticity. An alternative explanation is that the concerted mechanism exists because the intermediate is too unstable. That is, placing a negative charge next to a good leaving group is not feasible.
Because these reactions are inherently two-step operations but are “forced” to be concerted, a lot of them have hidden intermediates on the free energy surface. This is just as many in our community have predicted for a long time.