| [Related articles/posters: 027 122 037 ] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
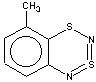
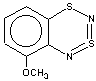
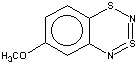
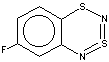
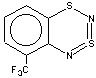
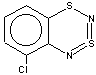
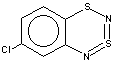
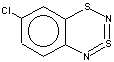
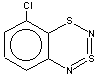
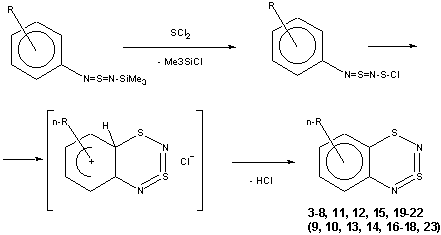
Thus, the target heterocycle formation proceeds smoothly with all the 2-RC6H4-N=S=N-SiMe3 (R=CH3, OCH3 , F, Cl, CF3) compounds tried to give corresponding 5-R-substituted derivatives of 1. In the case of 4-RC6H4-N=S=N-SiMe3 the cyclization readily occurs with R=CH3, Cl, while with R=OCH3 , F, CF3 and NO2 the expected 7-R-substituted derivatives of 1 are not found in the reaction mixtures. This is somewhat unexpected because the site of ring closure meta to R is the same for both 2-R- and 4-RC6H4-N=S=N-SiMe3.
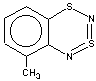
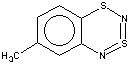
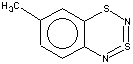
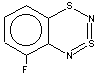
With 3-RC6H4-N=S=N-SiMe3 (R=CH3, OCH3 , F, Cl) the cyclization is highly regioselective leading exclusively (R=OCH3, F ) or predominantly (R=CH3, Cl ) to 6-R-substituted derivatives of 1; the ratio of the major 6-R isomer to the minor 8-R one is 78:22 (R = Cl) and 72:28 (R=CH3) as shown by 1H NMR spectroscopy (the structure of the 6-CH3 isomer 4 and of the 6-F isomer 12 has been confirmed by X-ray crystallography, see Figure 1). In the case of R=CF3 the cyclization fails, probably due to an unfavorable situation for electrophilic ring closure involving distribution of effective charges, qi, around the carbocycle perimeter.
The preferred direction of cyclization of 3-RC6H4-N=S=N-SiMe3 is consistent with the thermodynamics of corresponding Wheland type s-complexes (modeling a late transition state of the electrophilic aromatic substitution), as well as factors of kinetic control for orbital-controlled El-Nu interaction 8. Thus, according to the DHfo (PM3) data, the 6-R isomer of a s-complex is more stable than the 8-R one by ~ 3 ( R=CH3, Cl; 8-R isomer of a final product being observable) or by ~ 6 (R=OCH3 , F; 8-R isomer of a final heterocycle not being detectable) kcal mol -1. Under the orbital control the site of the cyclization is determined by ci2 distribution for the Nu's HOMO (taken as aromatic ring part of the HOMO (PM3) of the [3-RC6H4-N=S=N-S-Cl] intermediate with El's LUMO being an S-Cl s*-antibonding MO) with the c62 value exceeding that of c22 by a factor of 2.3 - 3.1.
According to the DHfo (PM3) data there is no specific destabilization of the corresponding s-complexes nor of the desired heterocycles in the case of the unsuccessful cyclizations as compared with the successful ones. It seems that a complex balance of different parameters of a molecular electronic structure (ci2 and qi distribution, ei values of Nu's HOMO and El's LUMO) is responsible for the cyclization failures observed.
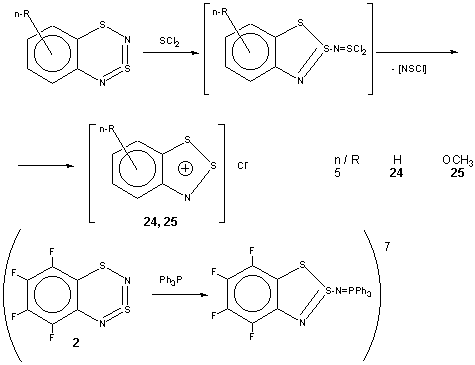
Cyclization By-Products. In some cases of both successful and unsuccessful cyclizations, the Herz salts (1,2,3-arenodithiazolium chlorides)9 have been isolated from the reaction mixtures (for selected examples, see Table 2) along with sulfur nitride (SN)4. Special experiments confirmed that it is possible to explain formation of the salts via interaction of the title compounds with SCl2 according to Scheme 3. The Scheme 3 is also substantiated by 14N NMR identification of thiazyl chloride NSCl (d14N 730 in CCl4) in the reaction mixtures. The identification was based on previously reported data19 as well as measurements on authentic sample prepared19 by heating a solution of (NSCl)320 in CCl4. At the same time, (NSCl)3 is not found in the reaction mixtures which corresponds to the conclusion that trimerization of NSCl is very kinetically hindered19.
Comparison of data presented above on the one hand, and data7
on the interaction of 2 with Ph3P (Scheme 3) on the other
hand, makes it possible to speculate that reaction of the title compounds
with formal both electrophile (SCl2) and nucleophile (Ph3P)
proceeds in a similar way, namely, as neutralization of Lewis acid (the
title compounds: low-lying vacant MOs; cf. Table 1)
by Lewis base (SCl2 and Ph3P: electron lone-pair).
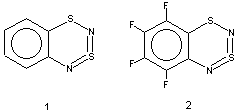
Molecular Structure. According to the X-ray diffraction data, the parent molecule 1 is planar 1, whereas heterocycle 2 is twisted along the S1 ... N4 line by 5.5o 2.
In the present work both types of molecular geometry were observed (Figure 1). In the case of derivatives of 1 possessing moderate or strong p-donor R substituents, 4 (R = 6-CH3) and 7 (R = 5-OCH3), the heterocycles were found to be bent along the S1 ... N4 line by ~ 7o (4) or ~ 11o (7). Contrary to 4 and 7, the heterocycles of derivatives of 1 with a weak p-donor and/or a strong s-acceptor substituent R, 12 (R = 6-F) and 15 (R = 5-CF3), are planar.
Figure 1. Molecular structure of compounds 4, 7, 12 and 15 (atomic coordinates, thermal parameters, bond lengths, and bond angles have been deposited at the Cambridge Crystallographic Data Centre). The bond lengths are typical1,2,18
The structural dichotomy found for the title compounds is reminiscent of the result previously observed for related 3,7-R2-1,5-dithia-2,4,6,8-tetrazocines: according to the X-ray diffraction data, with R = C6H5 the molecules are planar while with R = (CH3)2N they are folded along an S1 ... N5 axis10. The distortion was rationalized as being driven by a pseudo-Jahn-Teller instability in the p-system, namely, by p-donor-induced mixing of the high-lying p-HOMO with a low-lying virtual s-MO11. It seems, that the same explanation is also valid in the case of the title compounds too. For verification, quantum chemical calculations are planned for the future.
Heteroatom Reactivity. The extensive investigation of the title compounds' heteroatom reactivity is in progress. Only few results obtained are presented below just to provide the examples.
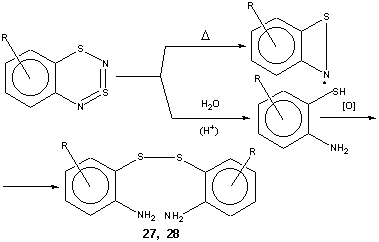
Thus, mild thermolysis of the title compounds (~ 150oC in squalane) leads to persistent radicals of benzo[c]-1,2-thiazet-yl type12 (Scheme 4) identified by ESR spectroscopy. Previously, only parent benzo[c]-1,2-thyazet-yl and its naphtho-analog, prepared in a different way, were known12. The thermolysis of the title compounds suggested here provides an easy access to the whole family of ring substituted benzo[c]-1,2-thyazet-yls.
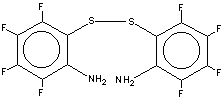
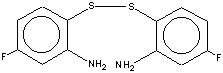
On mild acid hydrolysis the title compounds afford 2-aminobenzenethiols, identified for technical reasons in the form of corresponding disulfides (Scheme 4), which is useful adjunct to known methods. In particular, some previously unknown derivatives become available (for selected examples, see Table 3).
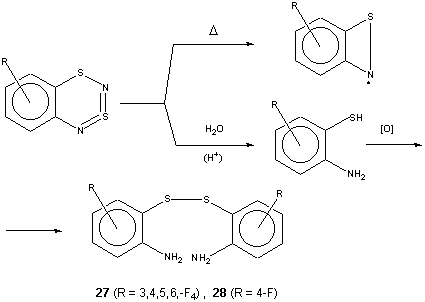
As the starting materials for the preparation of the title compounds are ArNH2 (à ArN=S=O à ArN=S=NSiMe3 à heterocycle) in hydrocarbon series (1 and this work), and ArFSH (à ArFSCl à ArFSN=S=NSiMe3 à heterocycle) in fluorocarbon series2, the formation of dithiadiazine followed by hydrolysis of the heterocycle can be considered to be a useful method for both ortho-thiolation of aromatic amines and ortho-amination of polyfluoroaromatic thiols (cf. also the related approach to ortho-amination of polyfluoroaromatic amines via polyfluorinated 2,1,3-benzothiadiazoles13).
For the reaction of the title compounds with SCl2 leading to the Herz salts, see Cyclization By-products (Scheme 3).
The X-ray structure determinations (Figure 1) were carried out on a Syntex P21 diffractometer using Cu-Ka radiation with a graphite monochromator. Corrections for both a systematic intensity drop and absorption were made. The structures were solved by direct methods using the SHELX-86 program and refined by the least-squares method in the full-matrix anisotropic (isotropic for H atoms) approximation using the SHELXL-93 and SHELXL-97 programs to R (wR2) 0.0475 (0.1252), 0.0501 (0.1381), 0.0633 (0.1593) and 0.0693 (0.1939) for 4, 7, 12 and 15, respectively. The PM3 calculations were performed with full geometry optimization.
The syntheses described subsequently were carried out, except for hydrolysis, in an argon atmosphere in absolute solvents with stirring.
Substituted 1,3l4d2,2,4-Benzodithiadiazines 3-8, 11, 12, 15, 19-22 (Table 1)
a) Solutions of 1.03 g (0.01 mol) of SCl2 and 0.01 mol of corresponding Ar-N=S=N-SiMe314, each in 30 ml of CH2Cl2, were slowly mixed by adding them dropwise to 300 ml of CH2Cl2 at 20oC, over a period of 1 h. After a further 1 h, the reaction solution was filtered, the solvent distilled off under reduced pressure, the residue sublimed in vacuo and the product recrystallized from hexane. Compounds 3-5, 7, 8, 11, 12, 15, 19-21 were isolated as black crystals. The precipitate was recrystallized from SOCl2:CCl4 (3:1) to obtain Herz salts.
In the case of Ar = 3-RC6H4, the filtrate was concentrated to an appropriate volume and the 1H NMR spectrum was measured to estimate the ratio of 6-R and 8-R isomers (enchanced shielding of H5,8 as compared with H6,7 makes it possible to assign unambiguously the signals of both isomers). After the same as above work-up of the solution, the major 6-R isomer was obtained by recrystallization while the minor 8-R isomer (6 and 22) was characterized only spectroscopically without eventual isolation.
b) In all other cases under the same conditions only (SN)4 and/or Herz salts were obtained along with some unidentified products.
Interaction of the title compounds with SCl2. Herz salts.
At 20oC, a solution of 0.31 g (0.003 mol) of SCl2 in 20 ml of CH2Cl2, was added during 45 min to a solution of 0.03 mol of one of the title compounds in 50 ml of the same solvent. After 15 min, the precipitate was filtered off, dried in vacuo, and recrystallized from SOCl2:CCl4 (3:1). Corresponding Herz salts9 were obtained as crystalline solids in nearly quantitative yields and characterized by high-resolution MS and multinuclear (1H, 13C and 14N) NMR (selected examples are given in Table 2).
To detect NSCl, the interaction was performed in CCl4 with more concentrated (~ 0.6 M) reactant solutions, and the 14N NMR spectrum of the reaction mixture was measured periodically during the reaction time.
Acid hydrolysis of the title compounds. 2,2'-Diaminodiaryl disulfides via 2-aminoarenethiols
At 0oC, to a solution of 0.02 mol of one of the title compounds in 10 ml of THF, was added a solution of 0.36 g of 1:10 diluted hydrochloric acid in 5 ml of THF. After 30 min, 15 ml of Et2O and then aqueous Na2CO3 were added. Organic layer was separated, dried over CaCl2, the solvent distilled off, and the residue recrystallized from hexane or heptane. 2,2'-Diaminodiaryl disulfides were obtained as (pale) yellow crystals (selected examples are given in Table 3).
Thermolysis of the title compounds. Ring substituted benzo[c]-1,2-thyazet-yls.
A 10-3 M solution of the title compound in squalane, degassed
by a series of three freeze-pump-thaw cycles, was gradually heated
in a ESR velve-equipped quartz capillary until ESR spectrum of a persistent
radical appeared (at ~ 150oC).
After 1 h, the sample was cooled to 20oC, diluted with hexane
(1:3), and ESR spectrum was measured. The ESR parameters of selected radicals
are given in Table 4. The simulated and experimental
spectra are in very good agreement.
Table 1. Substituted 1,3l4d2,2,4-Benzodithiadiazinesa
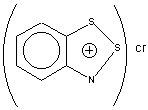
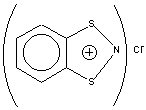
Table 2. Multinuclear NMR Data for the Herz
Saltsa 24, 25 and their isomeric 1,3,2-Benzodithiazolium Chloride
2615,16
Table 3. Novel Substituted 2,2'-Diaminodiphenyl
Disulfidesa Prepared from the Title Compounds
|
|
|
|
|
||
|
|
|
||||
|
|
 |
|
|
|
|
|
|
 |
|
|
|
|
Table 4. ESR Parameters of Radicals 29
and 30a, b
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.0078 |