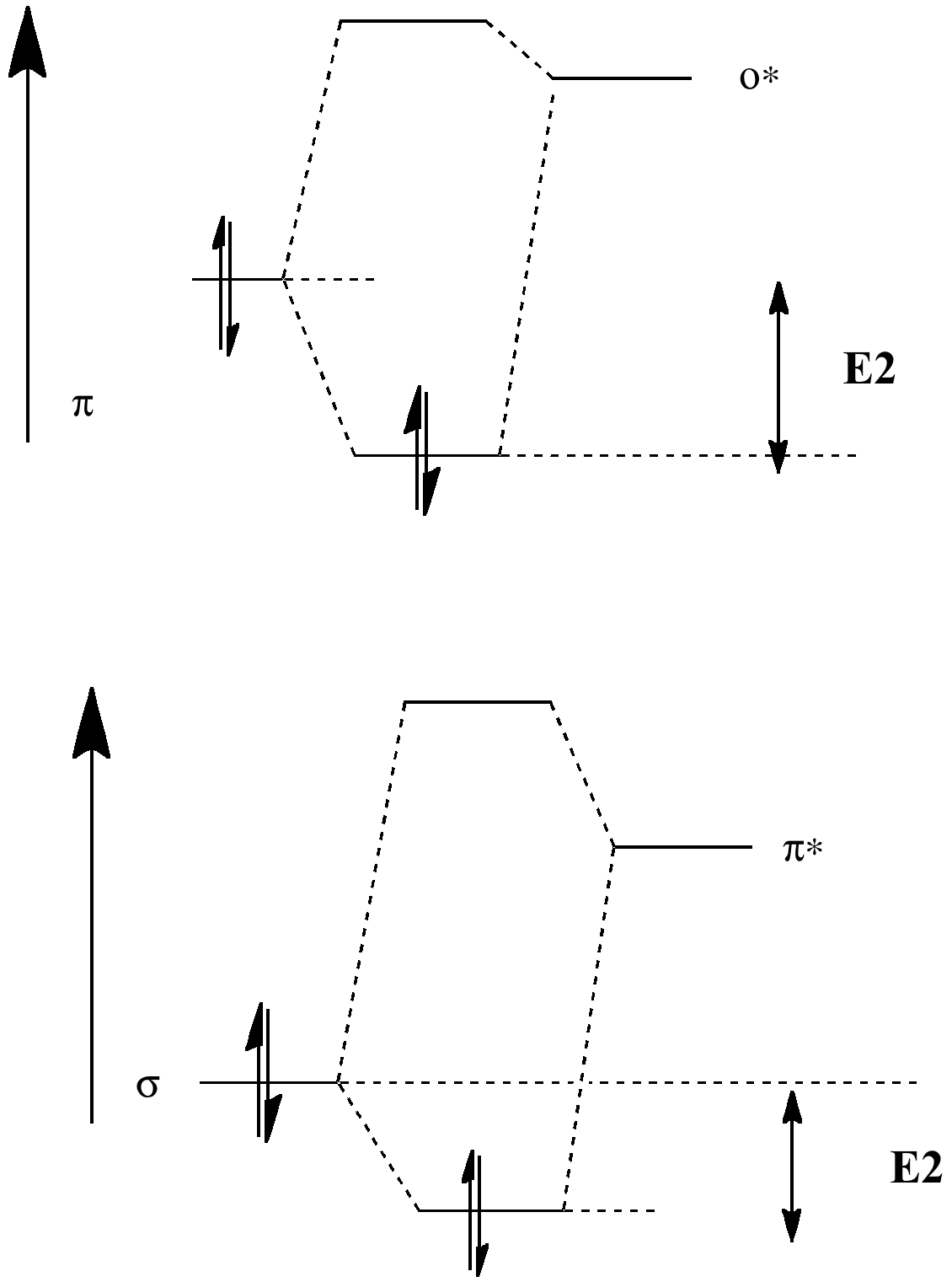
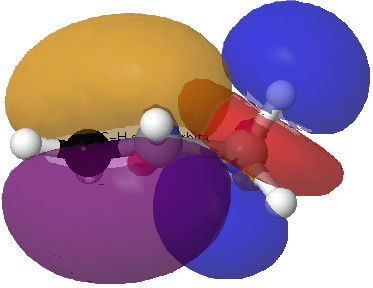
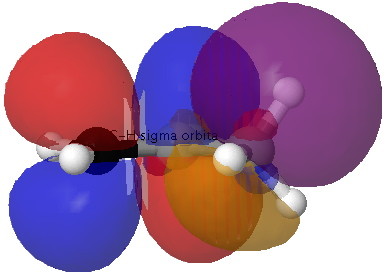
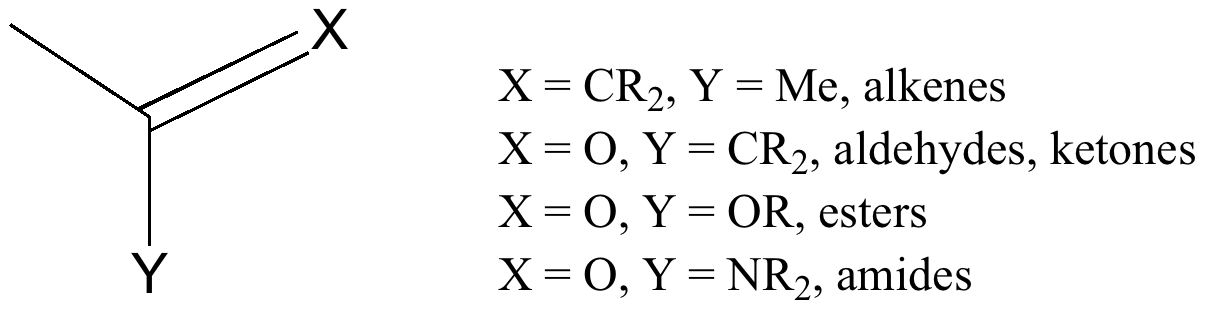
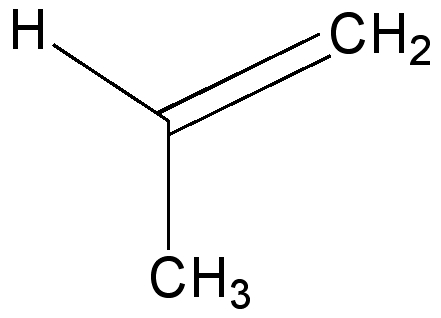
Propene is the simplest alkene containing such a bond. Rotation about the Csp2-Me bond is easier than the corresponding bond in propane for two reasons; there is one group fewer on the sp2 carbon and one of the bond angles is wider. This increases the distance between H and CH2 if they become eclipsed, and avoids the dreaded H...H contacts of < 2.1Å (Nos 2 and 3 in the previous lecture).
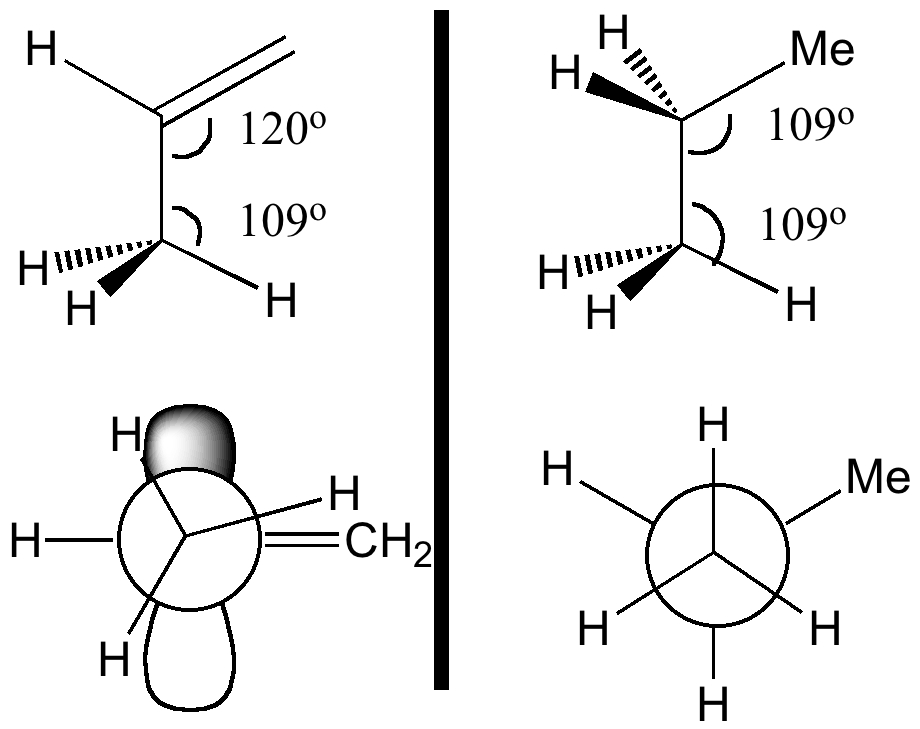
Orbital alignment also plays a part (previous lecture). Thus a π-bond has a small energy gap to the low energy π*-bond (i.e. a good acceptor, which is why carrots are orange). The reciprocal arrangement of πC=C/σ*C-H also happens to be favourable, since not only is the π*-bond low in energy, the π-bond itself is high in energy (the band gap is small, making the π bond also a good electron donor, and again why carrots are orange).
| πC=C/σ*C-H E2 = 3.6 kcal/mol | |
|---|---|
|
|
| σC-H/π*C=C E2 = 5.4 kcal/mol | |
|
Although the ideal orbital alignment might seem to be 90° to the π-system, is only possible for one C-H at any time, and in fact the system prefers to have two C-H bonds aligned at 120° (instead of just one at 90°), which allows both the "outward leaning" C-H bonds to effectively overlap with the π*C=C orbital (its antibonding nature makes it lean outwards as well). The result of these stabilising interactions is to force an eclipsing of one of the methyl hydrogens with the cis-hydrogen on the CH2 group:
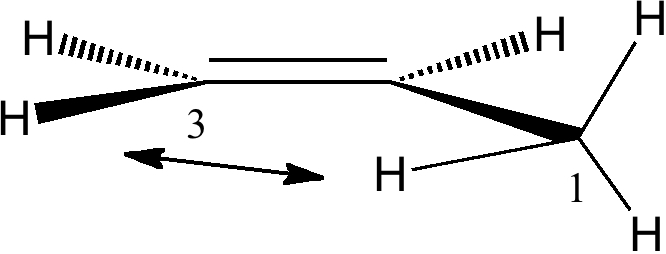
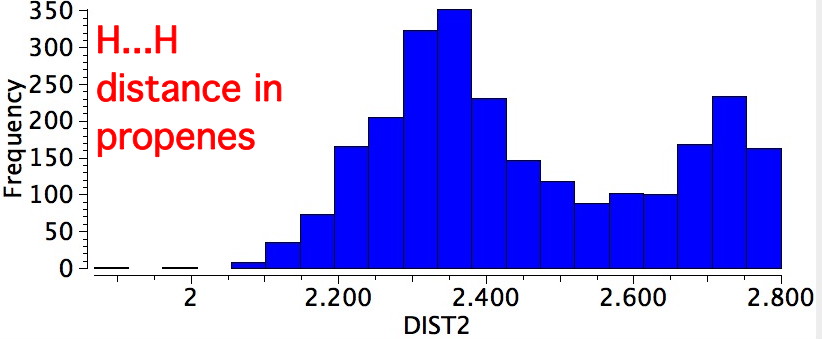
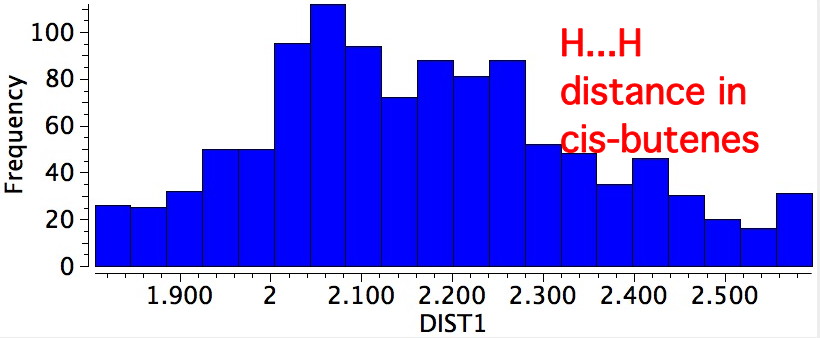
In fact, the H...H distance of this pair of hydrogens is ~2.47Å, slightly van der Waals attractive. This is called the A1,3 eclipsed conformation. We have what is called a win-win-win situation! The smaller peak at ~2.7Å is the alternative gauche conformation.
There is one special case of this which in fact you are already very familiar with; the (e.g. isopropyl, R=Me) carbocation.
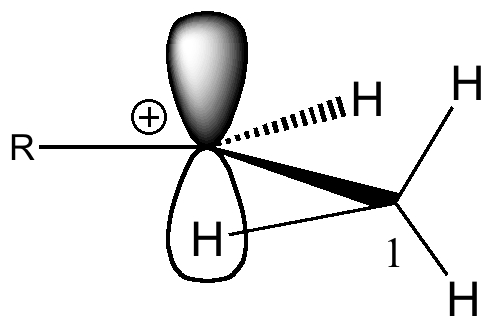
In this system, the low energy π*C=C NBO orbital is replaced by simply an empty p*π atomic orbital on one carbon. The +ve charge makes this orbital much lower in energy, and hence a far better electron acceptor than the π*C=C. Accordingly, the energy gap to the C-H σ-donor orbital is much smaller, making for a better interaction. The σC-H/p*π value for E2 is large: 41.6 kcal/mol. The H-H A1,3 eclipsed distance is 2.53Å.
These effects are in fact the electronic origin of the famous order of stability of carbocations that you will have seen in many other lecture courses.
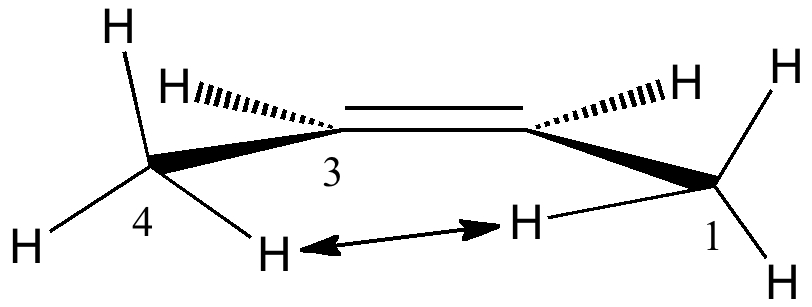
![]() This famously controversial system adopts a A1,4 eclipsed orientation (DOI: 10.3891/acta.chem.scand.24-0043) with a H...H distance of 2.1Å. The controversy is over whether the H...H interactions are in fact attractive or repulsive. The σC-H/π*C=C interaction remains significant (5.2 kcal/mol) and in fact is optimal in the A1,4 conformation, which allows the "outward leaning" π*C=C antibonding orbital to overlap effectively with similarly "outward leaning" C-H bonding orbitals
This famously controversial system adopts a A1,4 eclipsed orientation (DOI: 10.3891/acta.chem.scand.24-0043) with a H...H distance of 2.1Å. The controversy is over whether the H...H interactions are in fact attractive or repulsive. The σC-H/π*C=C interaction remains significant (5.2 kcal/mol) and in fact is optimal in the A1,4 conformation, which allows the "outward leaning" π*C=C antibonding orbital to overlap effectively with similarly "outward leaning" C-H bonding orbitals
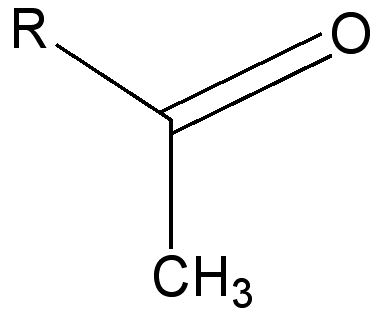
The σ-π* conjugation is greater for carbonyl double bonds than for alkene double bonds because the electronegativity of the oxygen atom lowers the energy of the π*C=O compared to π*C=C.
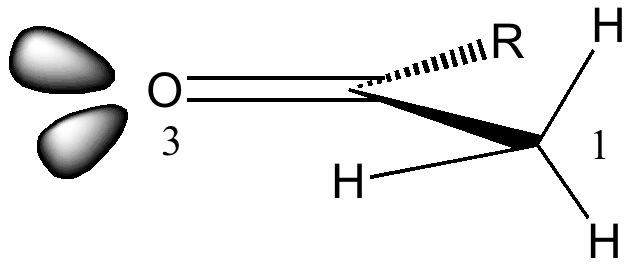
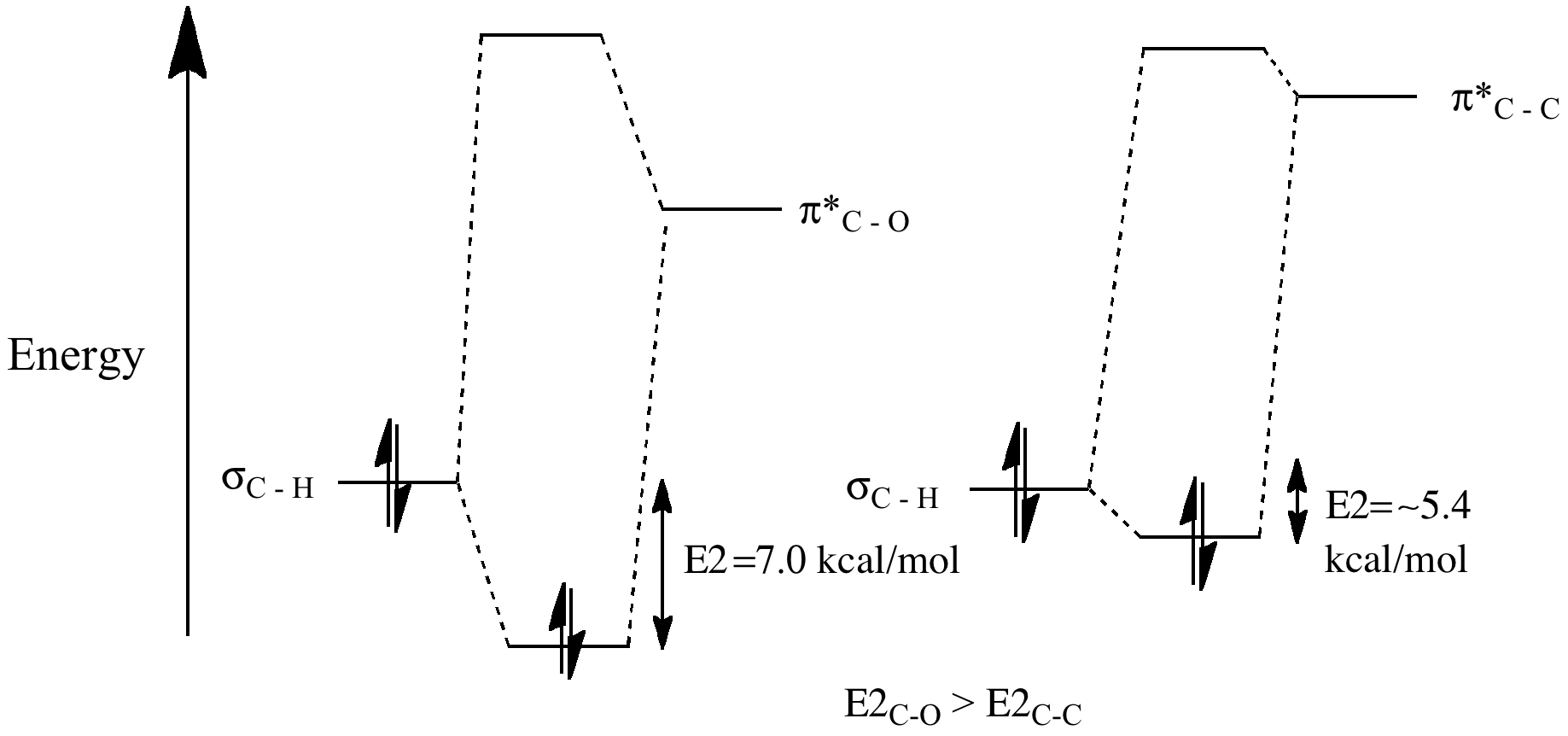
This increase in σ-π* stabilization is reflected in the relative pKa values for the α-protons of carbonyl compounds (16-20) compared to the allylic protons of alkenes (~43), since the greater interaction of the σC-H with the π*C=O (smaller σ-π* gap) will reduce the electron density in the C-H bond thereby making that proton more acidic.
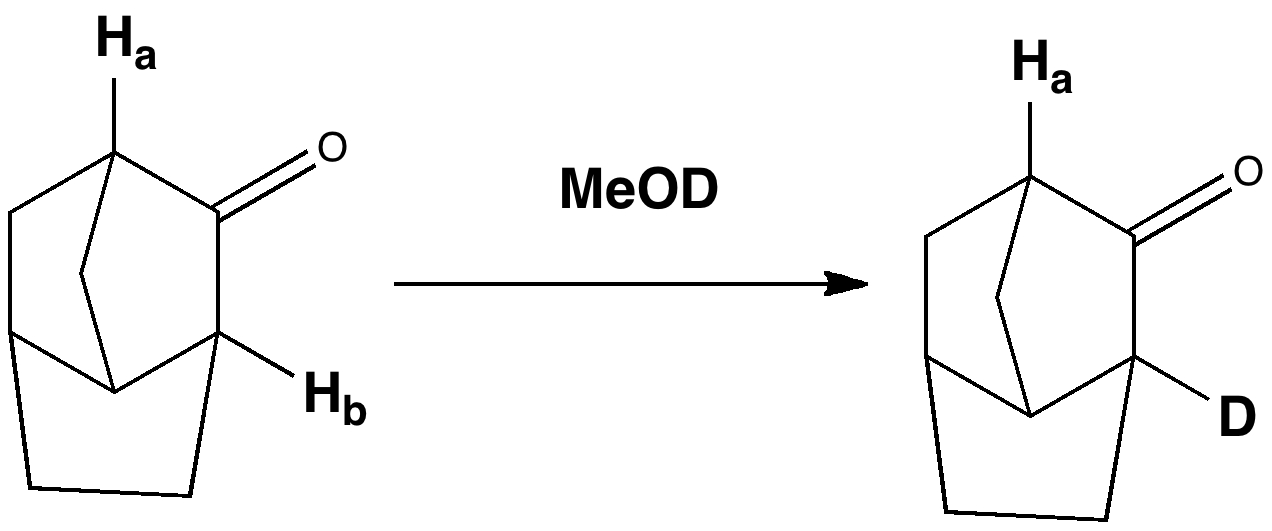
A good example of this effect is illustrated below. The cyclic nature of this ketone means that the orientation of the α-CH protons adjacent to the carbonyl is fixed and rigid. Only the proton marked Hb is replaced by deuterium when reacted with methanol-d1; Ha is untouched (DOI: 10.1021/ja00837a043). Thus σC-Hb/π*C=O is 5.4 kcal/mol, whereas σC-Ha/π*C=O is 0.5 kcal/mol. The greater σ-π* interaction corresponds to formation of H+, in other words it indicates how acidic the proton is. This acidity in turn arises because Hb is oriented ~60° to the C=O π-system, whereas Ha is only ~17° (i.e. more orthogonal and less capable of conjugation). This also manifests in the vibrational wavenumbers of the two C-H bonds. The less acidic one is (calculated) to be 3151 cm-1, whereas the more acidic one is 3102 cm-1.
The rotation barrier about the Me-Csp2 bond tends to be lower for carbonyl compounds (~1 kcal/mol) than for alkenes (~2 kcal/mol) despite the better σC-H/π* interaction, and that might be rationalised by the absence of any H...H van der Waals attraction in the 2.4Å region for the former (lone-pair...H interactions do not appear to generate such attraction).

Finally, σ-π* conjugation also explains why aldehydes are less stable (more reactive) than ketones since there are fewer α-protons in the former than in the latter and, hence, fewer possibilities for overlap.
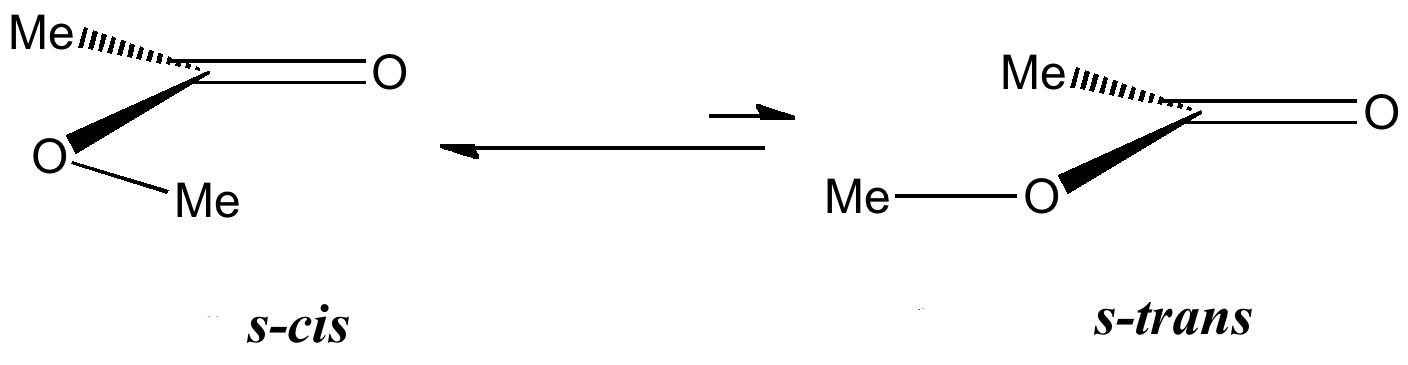
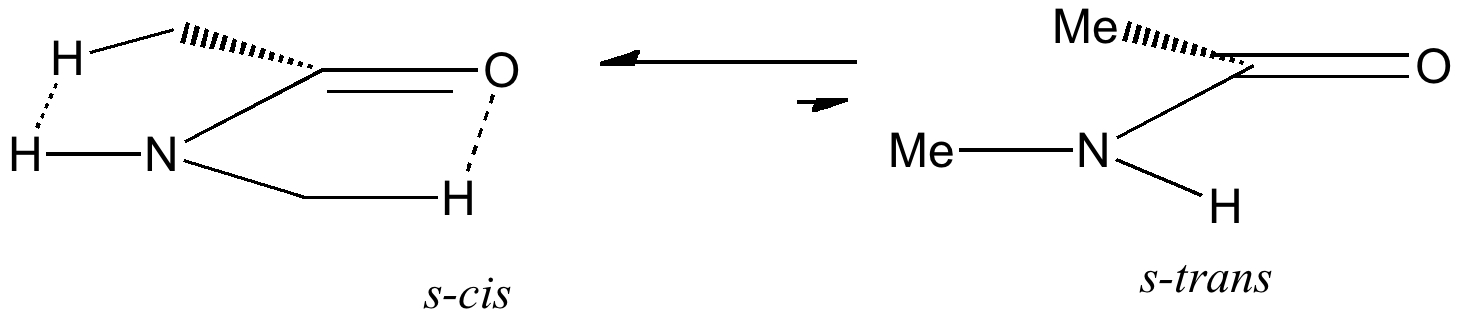
These can exist in two conformations or rotamers about the single bond, s-cis and s-trans.
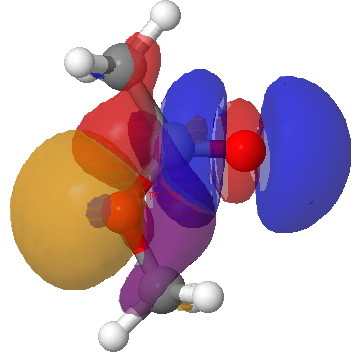
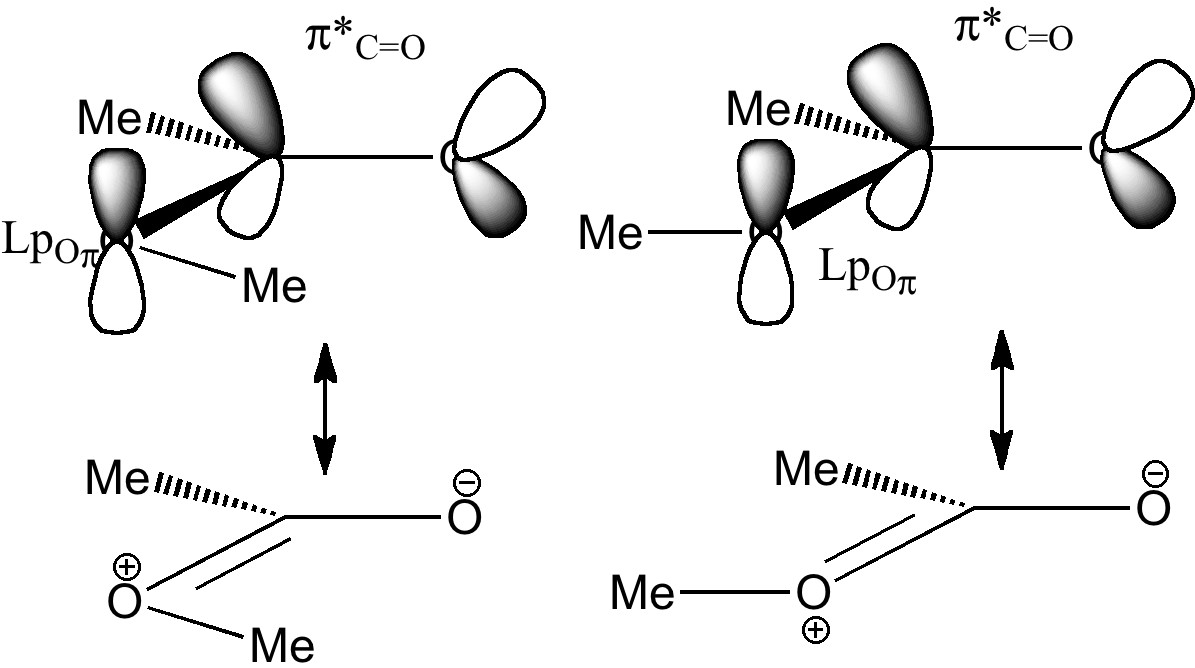
This introduces a new type of interaction, between an electron lone pair NBO orbital and either σ*C-O or π*C=O orbital. We have already seen how the π*C=O orbital is a good acceptor (low in energy). A lone pair (Lp) is an even better electron donor than a σ bond (it is only bound by one nucleus, not two), and hence not surprisingly this combination leads to large E(2) interactions.
This difference accounts for the preponderance of the s-cis conformation.
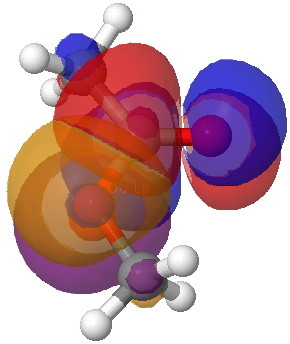
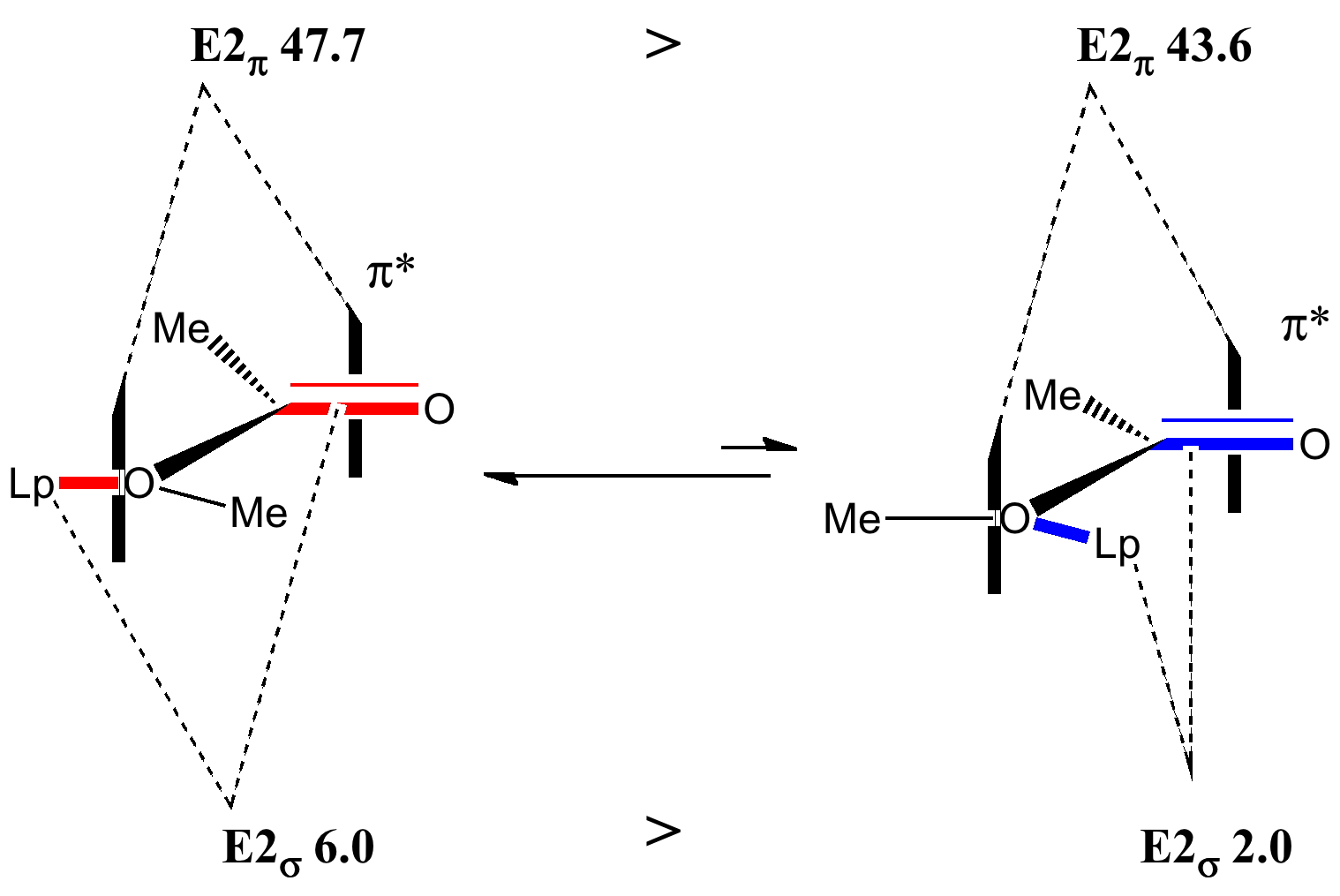
A far larger effect is E2 πOLp/π*C=O 47.7 kcal/mol for s-cis, which of course is pure π-π conjugation of the type you would have seen in e.g. benzene. The value for the s-trans isomer is 43.6 kcal/mol, lower than s-cis because the π* anti-bonding orbital "leans outward" and overlaps better with the s-cis lone pair than the s-trans.
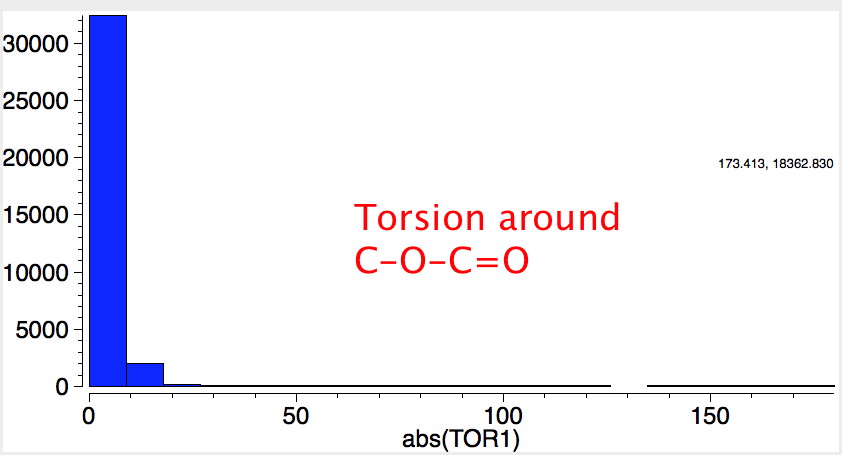
The partial π-double bond character of the O-C(=O) bond is reflected in a higher barrier to rotation about this bond (~12 kcal/mol), compared with the corresponding bond in methyl ethyl ketone at ~2 kcal/mol, although rotation is still fast at room temperature (we are still dealing with a conformational analysis rather than a configurational analysis!). The overall analysis can be summarised as:
![]() The balance between these two ester conformations can also be affected by the solvent. The dipole moment of the s-cis conformer is 1.8D, whereas the s-trans conformation is 4.1D. Polar solvents will preferentially stabilize the latter, whilist the former will be favoured in non-polar solvents.
The balance between these two ester conformations can also be affected by the solvent. The dipole moment of the s-cis conformer is 1.8D, whereas the s-trans conformation is 4.1D. Polar solvents will preferentially stabilize the latter, whilist the former will be favoured in non-polar solvents.

![]() This conformational preference in esters is critical to understanding why e.g. the Sharpless asymmetric epoxidation reaction is so enantioselective.
This conformational preference in esters is critical to understanding why e.g. the Sharpless asymmetric epoxidation reaction is so enantioselective.
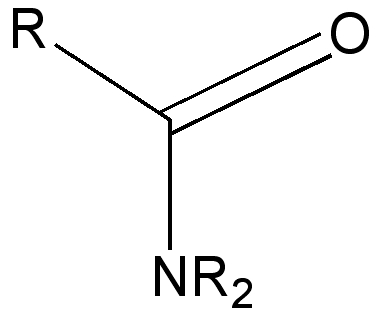
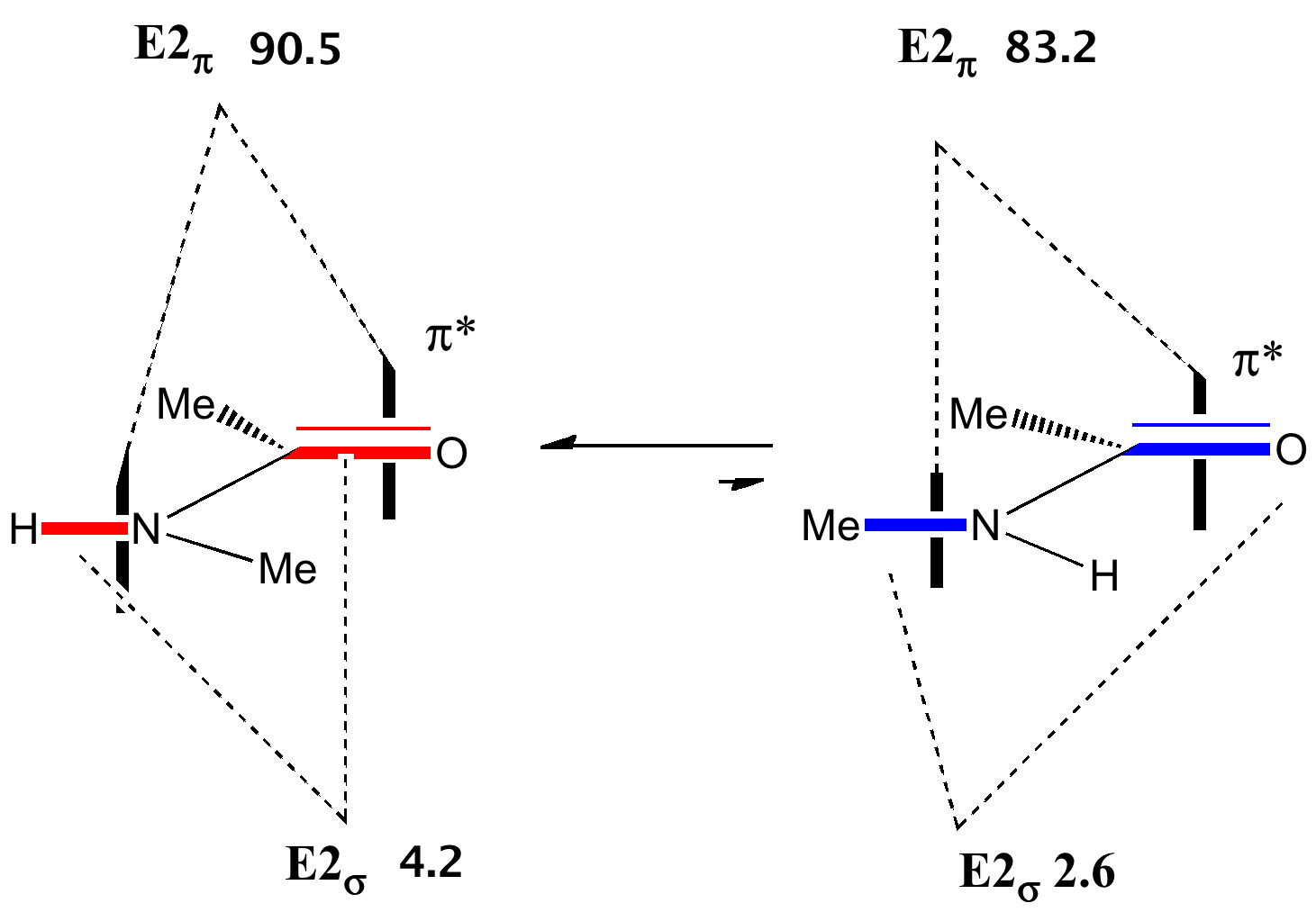
Amides continue the trend seen for esters. The πLp lone pair is an even better donor (higher in energy) than a Lp on an oxygen, because the nuclear charge on the N is smaller than O, and hence the lone pair is even less strongly bound by the nucleus. The σLp lone pair does not even exist for amides (it having been sequestered by one N-R).
Amides with an NH group do prefer to favour the s-cis form by ΔG 2.5 kcal/mol over the s-trans conformation
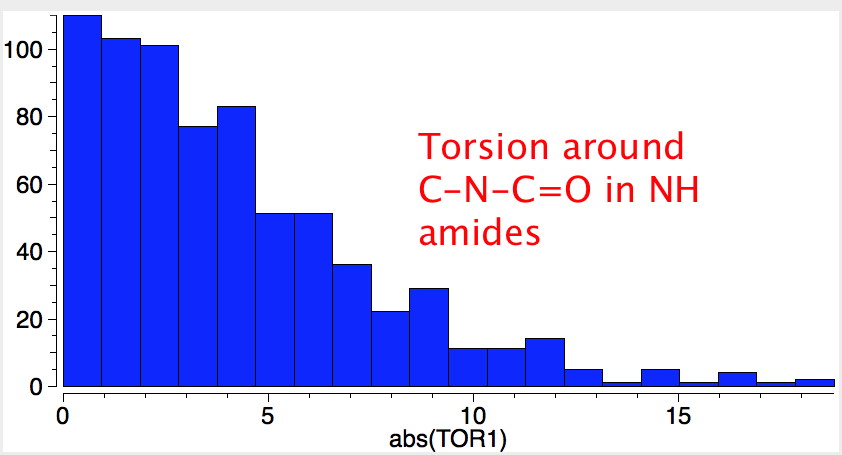
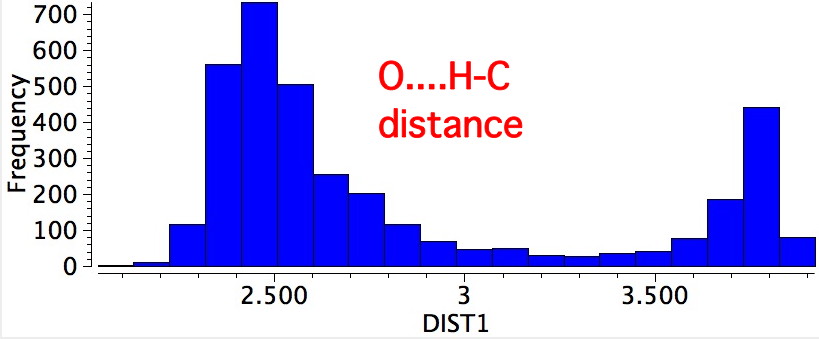
Both the π-π and σ-resonance favour the former, along with two short contacts (H...H 2.23Å, O...H 2.34Å). The second of these may be a weak hydrogen bond rather than a weakly attractive van der Waals effect (there being a continuum between these two descriptions).
The πNLp/π*C-O E2 interaction in amides is almost twice as large as for esters, and hence the rotational barrier about the N-C(=O) bond is also much higher (~18-20 kcal/mol).
This particular conformational effect it could be argued is the most important in all of biology since
it determines the structure of all proteins and enzymes. Put simply, this rotational barrier has
the appropriate value for life to evolve at ~37°C (a higher barrier would result in over-rigid proteins, a lower barrier
would result in over-floppy proteins) ![]()
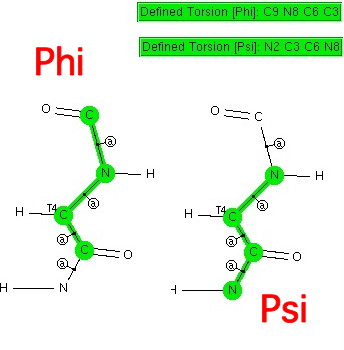
The s-cis conformation of the peptide bond also controls important aspects of the higher-order structure of poly-aminoacids (peptides), such as their propensity to form a right-handed α-helix. The conformational analysis of peptides is often represented in terms of two other important dihedral angles Phi and Psi about the backbone sp3 carbon linking two amide groups.
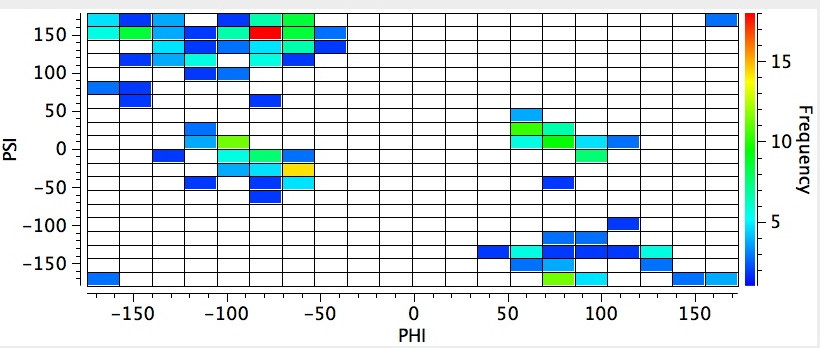
Historical note:![]() In 1951, Linus Pauling (DOI: 10.1073/pnas.37.4.205) used this information to propose a structure for how proteins fold.
He built models which were based on the s-cis conformation of amides and the planarity
resulting from the restricted rotation about the C=N bond of the peptide linkage, together with N-H...O=C hydrogen bonding.
These assumptions enabled him to reduce the complexity of the models to a solvable extent.
If you want to find out more, go read this gripping article.
In 1951, Linus Pauling (DOI: 10.1073/pnas.37.4.205) used this information to propose a structure for how proteins fold.
He built models which were based on the s-cis conformation of amides and the planarity
resulting from the restricted rotation about the C=N bond of the peptide linkage, together with N-H...O=C hydrogen bonding.
These assumptions enabled him to reduce the complexity of the models to a solvable extent.
If you want to find out more, go read this gripping article.
© Henry S. Rzepa, 2010-2014. Hide|show Toolbar.