X-ray structure of 29:
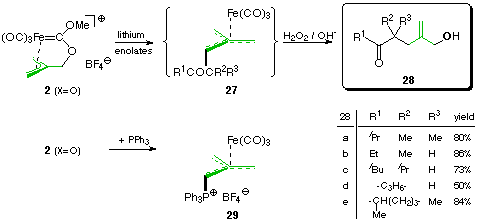
The carbene complex 2 (X=O) reacts with triphenylphosphane - just like its linearly tethered analogue 1 (X=O) (see path B)- with concomitant loss of carbon monoxide and methanol to give the substituted trimethylenemethane complex 29.
With carbon nucleophiles like lithium enolates, complex 2 reacts to give rather unstable intermediates which we have not been able to isolate and characterize so far and only tentatively ascribe the structure 27. When immediately treated with H2O2 / NaOH, these intermediates are oxidized to leave the corresponding allylic alcohols 28 in good yields. As the starting complex 2 itself derives from isobutene diol, this sequence makes for a convenient 3-step procedure for the monosubstitution of one hydroxy group by a beta-oxo-alkyl residue [3].
Experimental data
Tricarbonyl[eta4-(triphenylphosphonio)trimethylenemethane]iron(0) tetrafluoro borat (29): A solution of carbene complex 2 (3.40 g; 10.0 mmol) in CH3CN (50 ml) was treated with triphenylphosphane (2.62 g; 10.0 mmol) at room temperature and then stirred for 12 h. All volatile components were removed in vacuo and the residue thus obtained was purified by CC over silica gel. Pure 29 could be eluted with CH3CN / CH2Cl2 1:1, giving yellow crystals upon evaporation of the eluate; m.p. 178°C (decomp.); yield 4.44 g (8.2 mmol; 82%). Ð IR(KBr): 3075, 2970, 2060, 1990, 1975, 1430, 1045 cm-1. - 1H NMR (CD3CN, 400 MHz): 1.83 [dd, 4J (3-Hb/4-Ha) = 4.3, 2J (4-Ha/4-Hb) = 1.22 Hz, 1 H, 4-Ha], 2.31 [dd, 2J (4-Ha/4-Hb) = 1.22, 4J (1-H/4-Hb) = 2.44 Hz, 1 H,
4-Hb], 2.59 [d, 4J (3-Ha/P) = 10.7 Hz, 1 H, 3-Ha], 2.87 [d, 4J (3-Hb/4-Ha) = 4.3 Hz, 1 H, 3-Hb], 3.21 [dd, 2J (1-H/P) = 5.8, 4J (1-H/4-Hb) = 2.44 Hz, 1 H, 1-H], 7.72-7.88 (m, 15H, Har).Ê- 13C NMRÊ(CD3CN, 100.5 MHz): 47.60 [d, 1J (P/C-1) = 74.80 Hz, C-1], 57.22 (C-4), 60.24 [d, 3J (P/C-3) = 18.30 Hz, C-3], 110.68 (C-2), 121.40 [d, 1J (P/Cipso) = 88.50 Hz, Cipso], 131.20 [d, 2J (P/Cortho) = 12.20 Hz, Cortho], 135.00 [d, 3J (P/Cmeta) = 10.7 Hz, Cmeta], 136.20 [d, 4J (P/Cpara) = 3.0 Hz, Cpara], 208.55 and 208.75 and 209.9 (CO). - 31P NMR (CD3CN, 162 MHz, H3PO4 extern): 22.2. - MS (70 eV); m/z (%): 317 (63) [M+ - Fe(CO)3], 262 (100) [Ph3P+], 181 (78), 124 (58), 44 (69), 28 (36). Ð C25H20BF4FeO5P (542.1): calcd. C 55.39, H 3.72; found C 55.81, H 3.90. - Crystal structure: Clear, bright yellow single crystals were obtained by slowly cooling a solution of 29 in dichloromethane/diethyl ether 1:1 to 0°C; formula C25H20BClF4FeO3P, molar mass 584.00 g mol-1 (including CH2Cl2), crystal size 0.30 x 0.20 x 0.10 mm, a = 32.781(11), b = 7.928(2), c = 20.434(6) Å, a = 90°, b = 92.08(2)°, g = 90°, V = 5307(3) Å3, T = 293(2) K, dcalc= 1.462 g cm-3, Z = 8, monoclinic, space group C2/c.
6. Synthesis of carbamates 28 (General Procedure): Solutions of the required lithium enolates in THF were prepared from 1.1 mmol of the carbonyl compound and then added via a cannula into a slurry of the carbene complex 2 (340 mg; 1.0 mmol) in THF (5 ml) kept at -78°C. The entire mixture was stirred for 2 h at -78°C, then allowed to warm to room temperature and all volatile components were finally removed in vacuo. The resulting residue was quickly extracted with diethyl ether, the extracts were concentrated on an oil pump and then swiftly chromatographed over a short plug of silica (diethyl ether/petrol ether 1:2). The crude products 27 thus obtained were weighed and immediately redissolved in methanol (20 ml) and chilled with ice. H2O2 (6 ml of a 30% aqueous solution; 6.0 mmol) was added, whereupon the solutions turned brownish. Then solid sodium hydroxide (240 mg; 6.0 mmol) was added within 2 h and stirring was continued for another 2 h. Finally, the mixtures were extracted thrice with diethyl ether (100 ml) and the combined extracts were washed with saturated aqueous NH4Cl solution and dried over MgSO4. After
removal of the solvent on a rotary evaporator, the crude products were purified by CC (silica gel; diethyl ether/petrol ether 1:2).
6-Methylene-3-oxo-2,4,4-trimethyl-heptan-7-ol (28a):
IR(neat):3400, 3060, 2940, 2910, 2850, 1690, 1460, 1370, 1250, 1080 cm-1. - 1H NMR (CDCl3, 400 MHz): 1.06 [d, 3J (2-H/CH3) = 6.60 Hz, 6 H, 1-H, 2-CH3], 1.18 (s, 6 H, 4-CH3), 2.35 (s, 2 H, 5-H), 2.79 (s, 1 H, OH), 3.13 [sep, 3J (2-H/CH3) = 6.60 Hz, 1 H, 2-H], 3.98 (s, 2 H, 7-H), 4.81 and 5.09 (s each, 2 H, =CH2).-13C NMR (CDCl3, 100.5 MHz): 20.15 (C-1 and 2-CH3), 24.88 (4-CH3), 34.55 (C-2), 41.22 (C-5), 47.92 (C-4), 66.14
(C-7), 114.06 (=CH2), 145.85 (C-6), 219.13 (C-3). Ð MS (70 eV); m/z (%): 184 (3) [M+], 141 (12) [M+ Ð iPr], 111 (22), 95 (94), 71 (62), 43 (100) [iPr+].Ê- C12H20O2 (184.3): calcd. C 71.70, H 10.94; found C 71.63, H 10.87.

