| [Related articles/posters: 079 074 119 ] |
Stanislav Rćdla, Jitka Urbćnkovćb Petr Vćchalb, Jan Taimr
aResearch Institute for Pharmacy and Biochemistry, Kourimska 17, 13060 Prague 3, Czech Republic b Prague Institute of Chemical Technology, 166 28 Prague 6, Czech Republic
Abstract
Treatment of 2-hydroxybenzonitrile and 2-hydroxy-4,6-dimethoxybenzonitrile with phenacylbromides under alkaline conditions at room temperature provided the corresponding 2-benzoylmethoxybenzonitriles. Cyclization of these compounds to the corresponding 3-amino-2-benzoylbenzofurans was achieved under various conditions. 3-Amino-2-(2-fluorobenzoyl) benzofuran (5) was then cyclized into benzofuro[3,2-b]quinolin-10(5H)-one (2). Alkylation of this compound with iodomethane took place preferentially on the nitrogen atom giving 5-methylbenzofuro[3,2-b]quinolin-10(5H)-one (6) as a major product.
Key words: 3-aminobenzofurans; benzofuro[3,2-b]quinolin-10(5H)-ones
Introduction
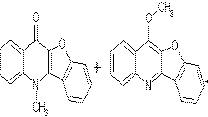
Recently Hradil and Jirman [1] described formation of 3-hydroxy-2-phenylquinolin-4(1H)-one (1) during an attempt to prepare 2-phenyl-3H-benz[e][1,4]oxazepin-5-one by treatment of anthranilic acid with phenacylbromide (Scheme 1).
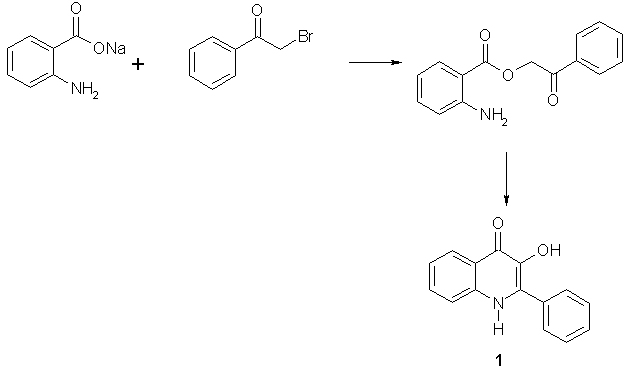
Scheme 1
They studied the reaction in detail and found it compatible with a wide range of substituents on both benzene rings [2]. The 2-phenylquinolin-4(1H)-ones are isosteric with naturally occurring flavones known for their biological activities. Therefore, it was not surprising that some of these compounds were found active in several tests. Molecular modeling revealed that the 2-phenyl group of 1 is only slightly out of the plane of the rest of the molecule. Therefore, we decided to study preparation of benzofuran[3,2-b]quinolin-10(5H)-one (2) as a rigid analog of these compounds.

A more instructive view of these two compounds is shown below.
Results and Discussion
There are some reports [3-5] describing synthesis of 2 and several its derivatives from suitable anthranilic acids and benzofuran-3(2H)-one or phenoxyacetic chloride (Scheme 2). In spite the ref. [4] described preparation of 2 in good yield using phenoxyacetic chloride, other authors [5] failed to reproduce the preparation and received only 15% yields. In our hands, the yield of this reaction was about 10-12 %. All other published modifications furnished compound 2 and/or its derivatives in yields too low for any practical applications, namely in the range of 0 to 16%. Therefore, we decided to prepare these compounds by a different way via corresponding benzo[b]furan intermediates.
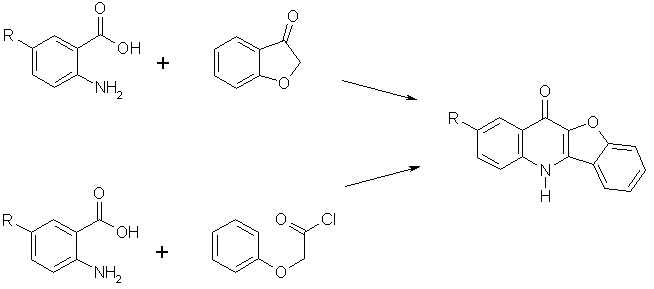
Scheme 2
Our convenient route for the synthesis of 2 started from 2-hydroxybenzonitrile 3 which is easily O-alkylated with a-haloketones to give the corresponding 2-acylmethoxybenzonitriles [6, 7]. We choose 4 as a suitable intermediate easily accessible from bromomethyl-(2-fluorophenyl)ketone. Similar 2-alkoxybenzonitriles having an acidic methylene group are known to cyclize under basic conditions to the corresponding 3-aminobenzofurans. We used sodium hydroxide at room temperature to get 4 which was after isolation cyclized with sodium ethanolate in ethanol at room temperature give good yield of 3-amino-2-(2-fluorobenzoyl)benzofuran (5). Further cyclization to 2 can be achieved either using NaH in N,N-dimethylformamide or, advantageously, refluxing a mixture of 5 and sodium methanolate in ethanol (Scheme 3).
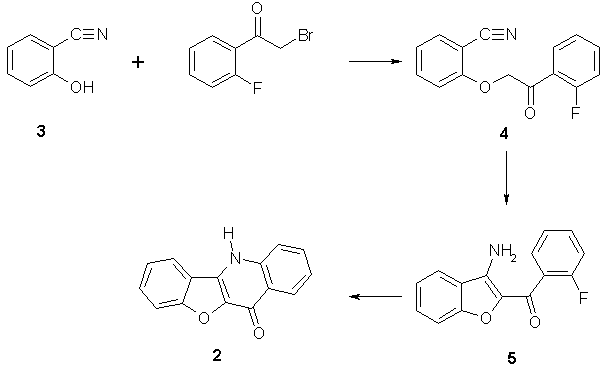
Scheme 3
Alkylation of 2 with iodomethane gave a mixture of major N-alkylation product 6 and minor O-alkylation product 7 suggesting that compound 2 is present mainly in its quinolone keto form (Scheme 4).
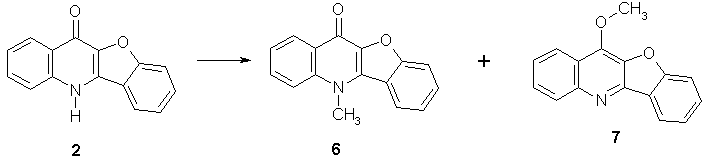
Scheme 4
Meanwhile high analgesic activity of benzofuran derivative 8 was published [8]. The compound is insoluble in water and therefore its potential therapeutic use is limited. Therefore we decided to prepare the corresponding 3-amino derivative 9 which could form water soluble salts.
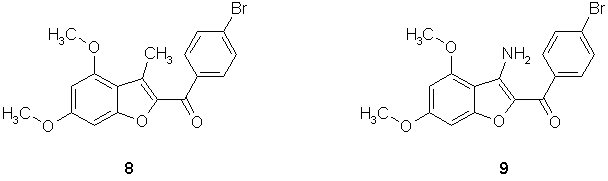
We used the same strategy described for the preparation of 5. Starting 2-hydroxy-4,6-dimethoxybenzonitrile (4,6-dimethoxysalicylonitrile) was prepared from commercially available 2-hydroxy-4,6-dimethoxybenzaldehyde via the corresponding oxime which was reduced in situ with formic acid to yield the benzonitrile derivative [9]. Alkylation of this compound with p-bromophenacylbromide in DMF in the presence of potassium carbonate provided the corresponding O-alkyl derivative 10, the cyclization of this compound into the corresponding 3-aminobenzofuran 9 was again achieved by sodium ethoxide or potassium t-butoxide at room temperature (Scheme 5).
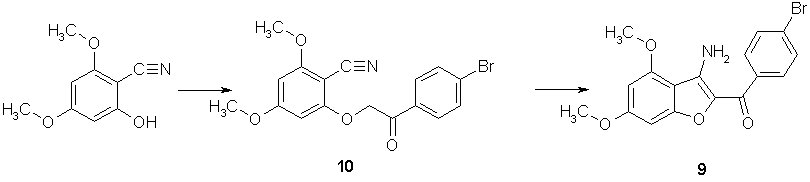
Scheme 5
Surprisingly, we failed to prepare any water soluble salt of this compound and the insoluble compound 9 itself was found to be inefficient as analgesics in several models.
Experimental
The melting points were determined on a Kofler block and are not corrected. The 1H and 13C-NMR spectra were measured on a Bruker DPX 250 spectrometer with tetramethylsilane as an internal standard. The IR spectra were scanned using a Shimadzu IR-445 spectrophotometer. UV spectra were taken on a Unicam 8800 spectrometer in ethanol. Mass spectra were measured on MCH 1320 and MAT 44S spectrometers. The purity of the substances prepared was evaluated by TLC on silica gel (FP KG 60 F254, Merck), preparative TLC was performed on pre-coated silica gel plates (PSC-FP KG 60 F254, Merck).
2-[(2-Fluorobenzoyl)methoxy]benzonitrile (4)
A solution of sodium hydroxide (0.6 g, 15 mmol) in water (1 ml) was added dropwise to a stirred solution of 2-hydroxybenzonitrile (1.9 g, 16 mmol) and 2-fluorophenacylbromide (3.5 g, 16 mmol) in 2-methoxyethanol and the mixture was refluxed for 20 minutes. Then the mixture was cooled down, the insoluble portion was filtered off, washed with a small amount of cold methanol and dried to give 1.9 g (50 %) of creamy crystals of m.p. 135-137 deg.C (ethanol). 1H-NMR (CDCl3) 5.37 (AB system, 2H, CH2), 6.79 (dd, 1H, J = 1.0 Hz, J = 8.5 Hz, H-3), 7.03 (d.t, 1H, J = 1.0 Hz, J = 7.5 Hz, H-5), 7.28 (ddd, 1H, J = 1.0 Hz, J = 8.2 Hz, J = 11.3 Hz, H-3Ō), 7.30 (m, 1H, H-5Ō), 7.47 (ddd, 1H, J = 1.6 Hz, J = 7.5 Hz, J = 8.5 Hz, H-4), 7.58 (dd, 1H, J = 1.6 Hz, J = 7.5 Hz, H-6), 7.62 (m, 1H, H-4Ō), 7.96 (d.t, 1H, J = 1.9 Hz, J = 7.2 Hz, H-6Ō). 19F-NMR (CDCl3) -108.31. MS, m/e 255 (M+, 100%), 254 (61%), 236 (55%), 235 (29%), 198 (8%), 160 (9%), 132 (8%), 123 (17%), 104 (21%), 95 (26%), 77 (31%), 51 (15%). Anal. Calcd for C15H10FNO2: C, 70.58; H, 3.95; F 7.44; N, 5.49. Found: C, 70.34; H, 4.04; F, 7.15; N, 5.49.
3-Amino-2-(2-fluorobenzoyl)benzofuran (5)
2-[(2-Fluorobenzoyl)methoxy]benzonitrile (4) (1.5 g, 6 mmol) was added during 1 h to a stirred solution of sodium (0.3 g, 13 mmol) in absolute ethanol (15 ml) and the mixture was stirredat room temperature for additional 1h. Then the mixture was poured in water (30 ml), the insoluble portion was filtered off, washecd with cold water and dried. Crystallization from ethanol provided 1.4 g (93 %) of yellow crystals, m.p. 162-163 deg.C. Anal. Calcd for C15H10FNO2: C, 70.58; H, 3.95; F 7.44; N, 5.49. Found: C, 70.14; H, 4.01; F; 7.08, N, 5.23.
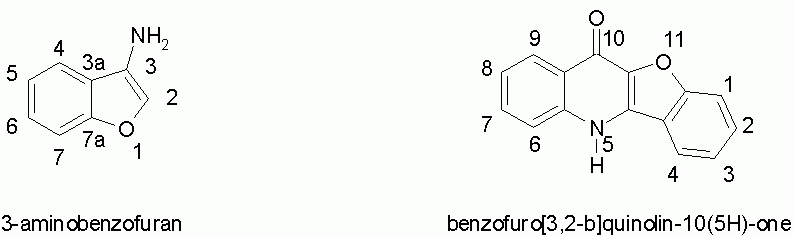
Benzofuro[3,2-b]quinolin-10(5H)-one (2)
Sodium hydride (50% suspension in mineral oil, 0.25 g, 5 mmol) was added to a stirred solution of 5 (0.5 g, 2 mmol) in DMF (10 ml) and the mixture was stirred for 2 h at 100 deg.C. The cold mixture was poured into water (50 ml), the insoluble portion was filtered off, washed with water and crystallized from methoxyethanol to give 0.42 g (91 %) of white crystals, not melting up to 360 deg.C. IR (KBr) 1530, 1580, 1645, and 2800 cm-1. UV (EtOH): lmax: 205, 217, 258, 307, 331, 345, 362 nm. 1H-NMR (DMSO-d6, 160 deg.C) 7.68 (m, 4H, H-1, H-2, H-6, H-7), 7.34 (m, 1H, H-8), 7.46 (m, 1H, H-3), 8.21 (bd, 1H, H-4), 8.40 (bd, 1H, H-9). 13C-NMR (DMSO-d6, 160 deg.C) 111.74 (C-1), 117.31 (C-6), 118.19 (C-4a), 120.77 (C-4), 120.85 (C-8), 122.33 (C-3), 124.64 (C-9), 125.29 (C-9a, C-10a), 128.83 (C-2), 130.29 (C-7), 137.31 (C-4b), 138.99 (C-5a), 154.73 (C-11a), and 164.60 (C-10). Anal. Calcd for C15H9NO2: C, 76.59; H, 3.86; N, 5.95. Found: C, 76.30; H, 4.07; N, 5.69.
5-Methylbenzofuro[3,2-b]quinolin-10(5H)-one (6) and 10-Methoxybenzofuro[3,2-b]quinoline (7)
Sodium hydride (50% suspension in mineral oil, 0.1g, 2 mmol) was added to a stirred solution of 2 (0.24 g, 1 mmol) in DMF (5 ml) and the mixture was stirred at room temperature for 1 h and then iodomethane (0.1 ml, 1.6 mmol) and the mixture was stirred for additional 10 h. Then the mixture was poured into water (25 ml), the insoluble portion was filtered off, washed with water and dried to give 0.31 g of crude mixture. The mixture was purified by preparative TLC using hexane - acetone 7 : 3 (v/v) to give 185 mg (74 %) of 6, not melting up to 360 deg.C (ethanol) and 35 mg (14 %) of 7, m.p. 117-119 deg.C (hexane). 5-Methylbenzofuro[3,2-b]quinolin-10(5H)-one (6) IR (KBr) 1508, 1585, and 1640 cm-1. UV (EtOH): lmax: 207, 220, 260, 310, 339, 354, 371 nm. 1H-NMR (DMSO-d6, 60 deg.C) 4.32 (s, 3H, CH3), 7.43-7.50 (m, 2H, H-3, H-8), 7.70 (m, 1H, H-2), 7.70 (m, 1H, H-2), 7.81 (m, 2H, H-1, H-7), 7.97 (bd, 1H, H-6) , 8.44 (bd, 2H, H-4, H-9). 13C-NMR (DMSO-d6, 60 deg.C) 37.57 (CH3), 114.84 (C-1), 117.95 (C-6), 120.88 (C-4a), 123.88 (C-8), 127.48 (C-9), 128.07 (C-9a), 128.28 (C-3), 128.58 (C-4), 131.63 (C-2), 133.72 (C-7), 136.65 (C-10a), 139.74 (C-5a), 142.50 (C-4b), 157.01 (C-11a), and 166.71 (C-10). Anal. Calcd for C16H11NO2: C, 77.10; H, 4.45; N, 5.62. Found: C, 76.66; H, 4.74; N, 5.60. 10-Methoxybenzofuro[3,2-b]quinoline (7) IR (KBr) 1198, 1575, and 1640 cm-1. UV (EtOH): lmax: 219, 253, 262, 287, 323, 335, 350 nm. 1H-NMR (CDCl3) 4.63 (s, 3H, CH3), 7.43 (bt, 1H, H-3), 7.56 (m, 3H, H-1, H-2, H-8), 7.68 (bt, 1H, H-7), 8.19 (d, 1H, J = 8.5 Hz, H-9), 8.33 (d, 1H, J = 8.5 Hz, H-6), 8.35 (d, 1H, J = 8.5 Hz, H-4). Anal. Calcd for C16H11NO2: C, 77.10; H, 4.45; N, 5.62. Found: C, 77.25; H, 4.66; N, 5.33.
2-Hydroxy-4,6-dimethoxybenzonitrile
A mixture of 4,6-dimethoxysalicylaldehyde (10 g, 55 mmol), hydroxylamine hydrochloride (4.4 g, 63 mmol), dry sodium acetate (4.5 g, 55 mmol), and anhydrous formic acid (40 ml) was refluxed under nitrogen for 3 h. The mixture was evaporated under reduced pressure, the residue was poured into water, the mixture was extracted with ether, the organic layer was washed with brine and dried with magnesium sulfate. The residue was purified by flash chromatography (petroleum ether - acetone 20 : 1) to give 3.35 g (33.5 %) of the starting aldehyde and 4.0 g (40 %) of white crystals, m.p. 203-206 deg.C (ethanol-water 1:1). 1H-NMR (CDCl3) 3.87 (s, 3H, CH3), 3.95 (s, 3H, CH3), 6.05 (d, 1H, J = 2.0 Hz, H-3 or H-5), 6.08 (d, 1H, J = 2.0 Hz, H-3 or H-5), 10.95 (bs, 1H,OH). 13C-NMR (CDCl3) 55.54 (CH3), 56.12 (CH3), 88.88 (C-1), 90.20 (C-5), 93.46 (C-3), 114.85 (CN), 162.52, 163.01, 164.73 (C-2, C-4, C-6). Anal. Calcd for C9H9NO3: C, 60.33; H, 5.07; N, 7.82. Found: C, 60.12; H, 5.28; N, 7.56.
2-[(4-Bromobenzoyl)methoxy]-4,6-dimethoxybenzonitrile (10)
A solution of 4-bromophenacylbromide (2.1 g, 7.5 mmol) in DMF (3 mL) was added dropwise under nitrogen to an ice-cooled mixture of 4,6-dimethoxysalicylonitrile (1 g, 5.6 mmol), dry potassium carbonate (1 g, 7.2 mmol) and DMF (10 mL) and the mixture was stirred at room temperature for 2h. Then the mixture was poured into water, the insoluble portion was filtered off, and crystallized from ethanol to give 2.1g (50 %) of white crystals of m.p. 190-191 deg.C. 1H-NMR (CDCl3): 3.79 (s, 3H, CH3), 3.87 (s, 3H, CH3), 5.26 (s, 2H,CH2), 5.96 (d, 1H, J = 2.2 Hz, H-3 or H-5), 6.08 (d, 1H, J = 2.2 Hz, H-3 or H-5), 7.65 (d, 2H, J = 8.5 Hz, H-3Ō, H-5Ō), 7.90 (d, 2H, J = 8.5 Hz, H-3Ō, H-5Ō). Anal. Calcd for C17H14BrNO4: C, 54.28; H, 3.75; Br, 21.24; N, 3.72. Found: C, 54.59; H, 3.84; Br, 20.85; N, 3.59.
3-Amino-2-(4-bromobenzoyl)-4,6-dimethoxybenzofuran (9)
Compound 10 (1.8 g, 4.8 mmol) was added during 1 h to a stirred solution of sodium (0.12 g, 5 mmol) in absolute ethanol (150 ml) and the mixture was stirred at room temperature for additional 1h. Then the mixture was poured in water (150 ml), the insoluble portion was filtered off, and crystallized from ethanol to yield 1.7 g (95 %) of yellow crystals, m.p. 225-226 deg.C. 1H-NMR (CDCl3): 3.84 (s, 3H, CH3), 3.92 (s, 3H, CH3), 6.20 (d, 1H, J = 1.9 Hz, H-5 or H-7), 6.42 (bs, 2H, NH2), 6.46 (d, 1H, J = 1.9 Hz, H-5 or H-7), 7.60 (d, 2H, J = 8.5 Hz, H-3Ō, H-5Ō), 8.06 (d, 2H, J = 8.5 Hz, H-3Ō, H-5Ō). Anal. Calcd for C17H14BrNO4: C, 54.28; H, 3.75; Br, 21.24; N, 3.72. Found: C, 54.56; H, 3.85; Br, 21.06; N, 3.66.
Acknowledgments
This study was supported by the Grant Agency of the Czech Republic (Grant No. 203/96/0112), and by Leciva Praha.
References
{kind=link}
{kind=link}