| [Related articles/posters: 077 026 118 ] |
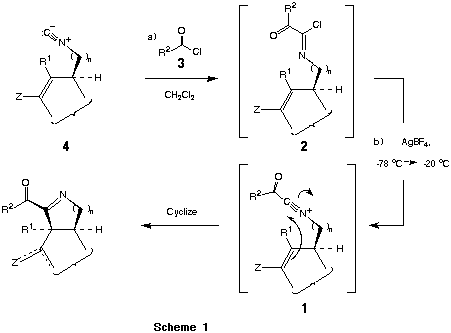
Cyclization reactions initiated by nitrogen-stabilized carbocations have continued to play a central role in synthetic approaches to numerous natural and unnatural heterocycles. Among the cationic species which have been employed for this purpose, iminium1 and acyliminium ions2 have proven particularly useful for effecting selective carbon-carbon bond formation. Analogous cyclizations initiated by nitrilium ions are considerably more obscure and often proceed inefficiently.3 The latter processes are frequently complicated by the methods used to generate the parent nitrilium ion and various side reactions (e.g., facile proton loss a- to the nitrilium moiety) that these intermediates undergo.
Recently, we required a general and exceedingly mild method for the construction of nitrogenous heterocycles of variable ring size. In principle, this objective could be met in a highly convergent manner by invoking the cyclization of a C-acylnitrilium ion 1 derived from an a-ketoimidoyl halide (e.g., 2) bearing an internal carbon nucleophile. These intermediates, in turn, were expected to be readily accessible by the reaction of an acyl halide 3 with the requisite isonitrile 4 (Scheme 1).
The synthetic advantages inherent to acylnitrilium ion initiated cyclizations include (1) a high level of convergence with respect to the introduction of peripheral 2-acyl moieties, (2) the flexibility to construct heterocycles of varied annular dimension, (3) the presence of an endocyclic imine within the product that can serve as a site for further functionalization, and (4) the exceptionally mild reaction conditions (AgBF4 or AgOTf, ClCH2CH2Cl/CH2Cl2, - 78® 0 oC) that are employed for effecting cyclization (eq 1).
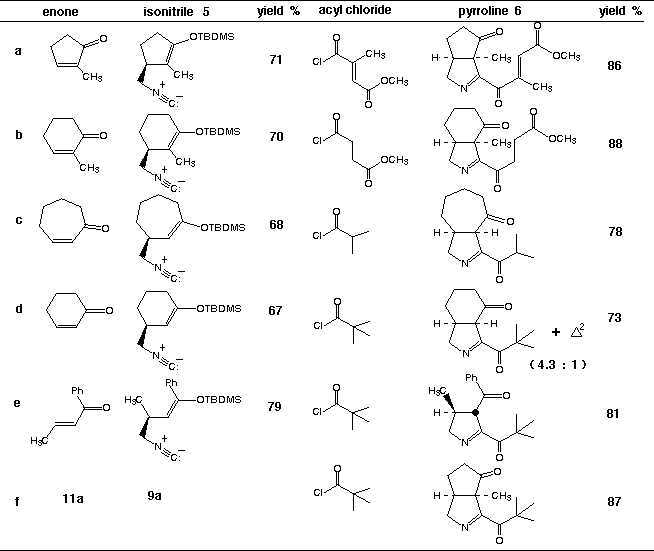
As part of a unified synthetic approach to the Orchidaceae alkaloids, we required large quantities of the D 1-pyrroline 6g. It was predicted that 6g and related D 1-pyrrolines might be synthesized in a highly convergent manner by the silver ion induced cyclization of a-ketoimidoyl chlorides (e.g., 8a-f) formed by the direct combination of acyl chlorides with isocyanomethylsilyl enol ethers (e.g., 9a-e). The requisite isocyanomethylsilyl enol ethers 9a-e, in turn, were expected to be available by sequential 1,4- addition of isocyanomethyllithium (10) to the corresponding a,B-unsaturated ketones 11a-e followed by enolate silylation. Curiously, there were no reports detailing the reactions of isocyanomethyllithium (10) or its organometallic derivatives with enones present in the literature at the time we began these investigations. Accordingly, we conducted a detailed study of the parameters which govern 1,2- vs 1,4- regioselectivity for this nucleophilic addition reaction. It was ultimately determined that reasonable to excellent selectivity favoring the desired 1,4- mode of addition could be achieved by simply complexing isocyanomethyllithium with TMEDA or HMPA prior to reaction with the enone. It is noteworthy that alternative organometallic derivatives of isocyanomethyllithium proved far less effective for 1,4-addition. It was subsequently found that 1,4- addition of complexed isocyanomethyllithium was extendable to a wide range of substrate enones. It was also discovered that the rate of reaction of the enolates derived from 1,4- addition with t-butyldimethylchlorosilane was significantly faster than the corresponding silylation of the tertiary alkoxides derived from 1,2- addition. Accordingly, simple hydrolysis of the reaction mixture resulting from sequential nucleophilic addition of LiCH2NC:, followed by silylation, provided the desired isocyanomethylsilyl enol ethers 9a-e admixed with small amounts of tertiary alcohols 12 which could be conveniently separated by flash chromatography. A compilation of the isocyanomethylsilyl enol ethers 9a-e which were prepared by this direct procedure appears in Table 1.
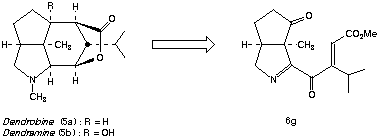
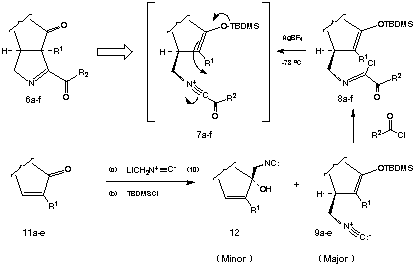
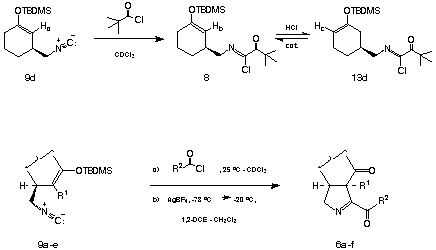
The acylative cyclization of the isocyanomethylsilyl enol ethers 9c and d initially appeared more problematic than the acylnitrilium ion initiated cyclizations of substrates bearing aryl terminators (vide supra).4,5 The reason for the inefficiency of cyclization was readily revealed by following the course of the reaction of 9d with trimethylacetyl chloride by 1H NMR. By way of this technique, the extent of acylation was easily monitored by following the disappearance of the signal attributable to CH2-NC: (d 3.14) and the development of the corresponding methylene signal associated with the a-ketoimidoyl chloride product 8d (d 3.45). During the course of acylation, the disappearance of the vinyl proton Ha (d 4.73) occurred with simultaneous development of a new signal attributable to Hb (d 4.69) and an unexpected vinyl resonance Hc assigned to the positional isomer 13d (d 4.71). The isomerization of 8d® 13d was presumably caused by trace amounts of adventitious HCl present in the reaction medium. It was readily determined that pyridine or, more conveniently, powdered 4 Å molecular sieves effectively suppressed silyl enol ether isomerization in substrates of this type. As expected, isocyanomethylsilyl enol ethers which possess tetrasubstituted alkene moieties were not found to undergo facile isomerization under the conditions which are typically employed (25 oC) to achieve isonitrile acylation.
The cyclization of the crude a-ketoimidoyl chlorides obtained in the above manner was achieved by their addition to 1.10 - 1.35 equiv of AgBF4 in CH2Cl2-ClCH2CH2Cl (1:1) at -78 oC followed by warming to -20 oC. An immediate precipitation of AgCl was observed at -78 oC suggesting the rapid generation of the transient acylnitrilium ion intermediates 7a-f. Subsequent cyclization of 7a-f occured at < -20 oC to afford the corresponding D 1-pyrrolines 6a-f in 85-95 % crude yield and in a high state of chemical purity as assessed by 1H NMR. The D 1-pyrrolines 6a-f prepared in this manner could be purified with modest recoveries when subjected to chromatography on untreated silica gel. However, the efficient purification of these compounds could be achieved either by reverse phase (C-18) chromatography or radial flash chromatography using silica gel disks that had been pretreated with gaseous Me3N. The results obtained from a series of D 1-pyrroline forming cyclizations are collected in Table 1.
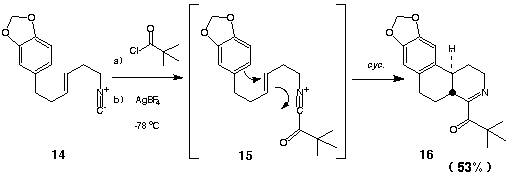
We had previously shown that acylnitrilium ions are sufficiently electrophilic to undergo facile cyclization with nonactivated arenes at -20 oC (vide supra).5 The use of simple alkenes as nucleophilic addends in acylnitrilium ion initiated heteroannulations would be of considerable synthetic interest. To explore this possibility, the unsaturated isonitrile 14 was sequentially acylated ((CH3)3CCOCl, 25 oC, 6 h) and then subjected to AgBF4-mediated cyclization (CH2Cl2-CH3NO2, -78 oC). As had been desired, the tetrahydropyridine 16 was obtained as the major cyclized product in 53% isolated yield. The extension of this methodology to the synthesis of naturally occurring ring systems was investigated in a later study (see part iii).
The examples presented above clearly indicate the utility of acylnitrilium ion initiated cyclizations for the synthesis of polyfunctional azacycles. The application of this methodology to the synthesis of the Orchidaceae alkaloids was subsequently undertaken.
Alkaloids belonging to the Lycopodium family have remained an enduring challenge for efficient chemical synthesis. Although a number of these alkaloids possess intriguing pharmacology, the ongoing interest in total synthesis would appear to be stimulated at least as much by the structural complexity of these substances and the attendant strategic imperatives for skeletal construction. Despite the continuing activity in this area, relatively little progress has been reported with regard to the synthesis of the irregular alkaloids belonging to the serratinane subgroup. With the exception of (±)-serratinine 17 and the corresponding 8-deoxy derivative, each of which have been synthesized once previously, no completed syntheses of alkaloids in this category have appeared.
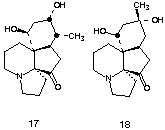
As described above, we had previously shown that cyclizations initiated by acylnitrilium ions could provide access to a wide variety of heterocyclic systems. Our interest in (±)-serratine 18 was stimulated by the possibility that the essential tricyclic core of this Lycopodium alkaloid could be derived from a simple arene nucleus via sequential utilization of an acylnitrilium ion initiated spiroannulation and intramolecular 1,4-addition (Scheme 2). Below we detail the successful implementation of this strategy.
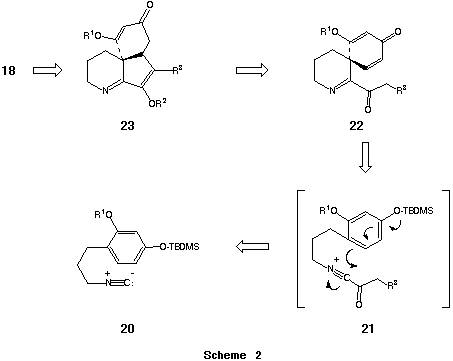
The key isonitrile 20a that was employed in this investigation was prepared by sequential formylation/dehydration (i. EtOCHO; ii. POCl3-Et3N, THF, 0 oC) of 3-[-4-(tert- butyldimethylsiloxy)-2-methoxyphenyl]propanamine 19. Acylation of 20a with the acyl chlorides 24a-f (CH2Cl2, rt) was found to proceed in close analogy with previous examples to provide the intermediate a-ketoimidoyl chlorides 25a-f in ca. quantitative yield (NMR). Exposure of 25a-f to AgBF4 (1.5 equiv) in ClCH2CH2Cl-CH2Cl2 (1:1) at 70 oC resulted, without exception, in the immediate precipitation of AgCl signalling the generation of the corresponding transient acylnitrilium ions. In contrast to most of the examples previously described in this review, cation interception by the pendant carbon centered nucleophile did not occur rapidly at 78 oC. Optimally, preformed solutions of the reactive intermediates were subsequently warmed to 20 oC and maintained at this temperature for 20 h to induce spirocyclization. By way of this procedure the spiro[cyclohexa-2,5-diene-1,3'-(3',4',5',6'-tetrahydropyridin)]-ones 22a-e could be obtained on a preparative scale in 80-84% purified yield. It is of interest that cyclization of 25f under an analogous set of reaction conditions resulted in only a modest yield (55%) of the desired spirocyclic intermediate 22f. Presumably, the enhanced propensity of the B-ketosulfonyl moiety of 25f to enolize was responsible for the degradation of spirocyclization efficiency in this instance. In this connection, the successful conversion of 25b-f to 22b-f constitutes the first reasonably comprehensive series of acylnitrilium ion cyclizations involving substrates that possess readily enolizable sites a- to the carbonyl function.
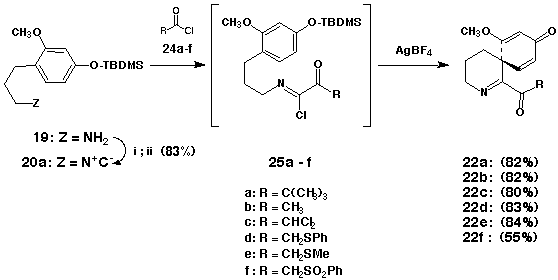
The predisposition of the spirocyclic intermediates 22b-e to undergo intramolecular 1,4-addition was subsequently examined. Several attempts to convert 22c to the corresponding tricycle 26a via free-radical intermediates under reductive conditions (Bu3SnH) led only to the production of 22b albeit in excellent (>90%) yield. By way of contrast, base-mediated cyclization of 22d and 22e gave more encouraging results. In an initial experiment, exposure of 22d to NaH (1.1 equiv) in DMF [0 oC (10 min)® 25 oC (8 h)] followed by protonolysis (AcOH, 1.1 equiv) afforded the unstable a-phenylthioketone 26b in good yield. We suspected that the observed instability of 26b might be a consequence of initial tautomerization involving the sensitive a-keto imine moiety. Accordingly, this bifunctional array was derivatized by alkylative protection in situ. To this end, cyclization of 22d (NaH-DMF, 0 oC® 25 oC) followed by enolate interception [SEM-Cl (1.1 equiv), 60 oC® 25 oC] furnished the tricyclic imine 23d in 96% purified yield. Cyclization of 22e in a similar manner provided 23e in 98% yield.48 Although 22f could be induced to undergo an analogous cyclization, 22b proved resistant toward base mediated intramolecular Michael addition.
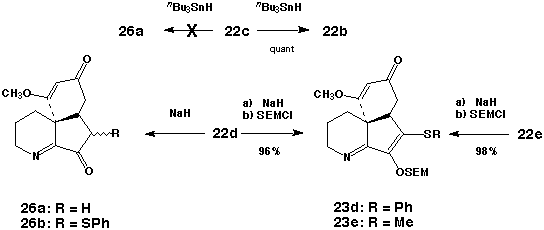
At least three features of the preceding cyclizations are worthy of comment. These studies have shown that a variety of functionally varied a-keto imidoyl chlorides can serve as effective precursors to synthetically viable acylnitrilium ions; intramolecular capture of the enolates derived from 22d and 22e by the least substituted (and most electrophilic) cyclohexadienone B-carbon is completely selective; and the in situ enolate trapping protocol gives rise to the functionally well differentiated intermediates 23d and 23e. Studies directed toward the stereodefined annulation of the serratinane D ring and the completion of the synthesis of (±)-serratine (18) are continuing.
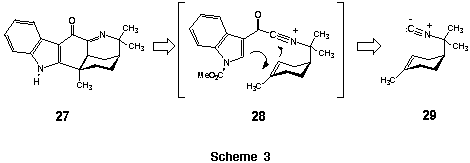
Our interest in the prospective use of an acylnitrilium ion initiated alkene-cascade cyclization for the synthesis of the Aristotelia alkaloid (+)-makonine (27) (Scheme 3) prompted our examination of prototypical cyclizations between these cations and simple, unactivated alkenes. The results of this study are described below.
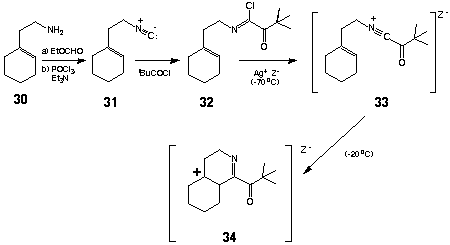
The isonitriles employed in this investigation were prepared from the corresponding amines by sequential N-formylation/dehydration. Accordingly, formylation of amine 30 (EtOCHO, reflux, 5 h) followed by dehydration (POCl3-Et3N, THF, 0 oC) furnished isocyanide 31 in 82% yield after distillation.
Cyclization Studies.
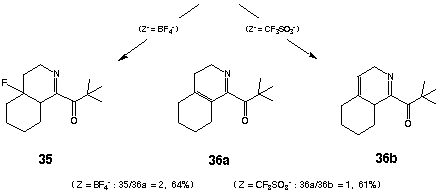
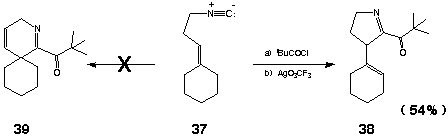
The initial conversion of compound 31 into the corresponding a-keto imidoyl chloride 32 was achieved by treatment with trimethylacetyl chloride (TMAC) (1.05 equiv) as described previously. Whereas ionization of 32 could be readily achieved by exposure to AgBF4 (1.10 equiv) in analogy with previous examples [CH2Cl2 (CH2Cl)2, - 70 oC], subsequent cyclization of the resulting acylnitrilium ion 33 took an unexpected course. When the reaction mixture was warmed to - 20 oC, cyclization of 33 proceeded by way of the tertiary carbonium ion 34 to provide 35 and 36a (35/36a = 2) in 64% isolated yield. The formation of 35 could be completely suppressed by the use of AgO3SCF3 for generation of the initial acylnitrilium cation. Accordingly, treatment of 32 with AgO3SCF3 (1.10 equiv, CH2Cl2, - 70 oC ® - 40 oC) provided the isomeric hexahydroisoquinolines 36a and 36b [36a/36b = 1 (NMR)] in 61% overall yield from the isocyanide 31. Exposure of this mixture to silica gel resulted in quantitative conversion of 36b into 36a which could be isolated in analytically pure form in 61% yield. As expected, the regiochemistry of the foregoing cyclizations (fused vs spiro) would appear to be governed by the relative stabilities of the respective post-cyclization carbocations. A similar outcome was observed in the acylative cyclization of 3-cyclohexylidene-1-isocyanopropane (37). Sequential reaction of 37 with TMAC (1.05 equiv) followed by ionization/cyclization of the resulting a-ketoimidoyl chloride [AgO3SCF3 (1.10 equiv), CH2Cl2,- 70 oC® - 40 oC] provided the 3,4- dihydro- 2 H- pyrrole 38 to the exclusion of the corresponding spirocycle 39 in 54% yield after chromatography. Not surprisingly, conformational restriction of the 3-(1-cyclohexenyl) substituent precluded facile isomerization of the nonconjugated alkene within 38 (vide supra).
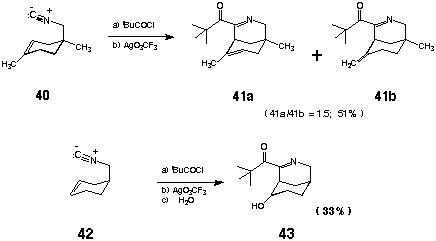
As a prelude to the intended synthesis of (+)-makonine (27), we next turned our attention to the construction of bicyclic ring systems via acylnitrilium ion-alkene cyclizations. Reaction of 4-(isocyanomethyl)-2,4-dimethyl-1-cyclohexene (40) with TMAC (1.05 equiv) followed by Ag+ mediated cyclization [AgO3SCF3 (1.10 equiv), CH2Cl2,- 70 oC® - 40 oC] secured the azabicyclo[3á3á1]-nonanes 41a and 41b [41a/41b = 1.5 (NMR)] in 51% yield after chromatography. Presumably 41a and b arise as a consequence of divergent proton elimination from a common carbocationic intermediate. In contrast to this result, acylation/cyclization of the 1,2-disubstituted alkene containing isonitrile 42 (vide supra) furnished the bicyclic alcohol 43 as the only major product after an aqueous quench albeit in low (33%) isolated yield. In this instance, a series of DEPT and 1H-13C HETCOR experiments was used to unambiguously establish the connectivity of the core heterocycle and rule out the isomeric [3á2á2] ring system. The regioselectivity of cyclization in the case of 43 is likely a reflection of preferential 6-membered ring formation.
The preceding examples clearly show that even isolated, unactivated alkenes can serve as suitable terminators for cyclizations involving acylnitrilium ions. Although cyclization efficiencies are lower than in previous examples, these results indicate that isolated alkenes in various settings might serve as effective cationic relay moieties for cascade type annulations.