| [Related articles/posters: 007 034 118 ] |
Abstract
The cycloaddition ligand delivery reagents 7,8-diazaphencyclone
9 and 3,6-di-(2'-pyridyl)-s-tetrazine 10,
were reacted with pre-prepared porphyrin dienophiles, to obtain
structurally organised porphyrin-ligand couples I-III.
Central to this process was the synthesis of rigid alicyclic
BLOCKs, which contained an a-dione
and a strained dienophilic p-centre, thereby providing the ability
to fuse the porphyrin (via porphyrin diamine 4a) and ligand moieties (by cycloaddition) site specifically onto the spacer framework.
The positioning of photochemically active moieties within larger
supramolecular systems has been a topic of increasing interest
in recent times. This has seen the development of a wide range
of systems designed for use in artificial photosynthetic studies,
molecular switches, or as molecular wires[1,2]. Within these
systems, the use of polypyridyl complexes of d6
metals including Ru(II), Os(II) and Re(I) has been widespread
due to their favourable photochemical character and well developed
synthetic chemistry[3]. Similarly, the investigation of the photochemical
and electrochemical properties of porphyrins has been studied[4].
However, in the vast majority of such studies, the combination
of porphyrins and polypyridyl complexes into a single structure
has remained surprisingly uncommon. Relevant studies include
the work of Hamilton[5], Sauvage and co-workers[6,7], and Crossley[8].
Examples of porphyrin-ligand structures from these studies are
illustrated in Figure 1.
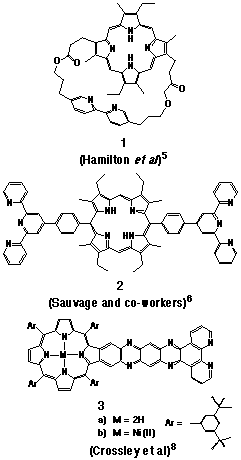
While the use of 3 as a molecular wire for the attachment of porphyrins to a gold surface has been alluded to, the application of Crossley's work appears to have been limited to the preparation of supramolecular species based upon square planer complexes of 3.[8] Photophysical studies on the ruthenium(II)-bipyridyl derivative of 1 has been reported and the extinguishment of the ruthenium-based luminescence by the porphyrin free base noted[5], although no mechanism for this quenching effect was discussed by the author. Similar studies by Sauvage and co-workers with the metal complex derivatives of 2 and other examples reported a similar effect, which was attributed to intramolecular triplet energy transfer from the ruthenium(II)-terpyridyl centre to the appended porphyrin[5]. The authors also reported in that study that intramolecular electron-transfer occurred from the photo-excited porphyrin to the attached ruthenium(II) or rhodium(III) complexes, an observation not reported by Hamilton.
Much of the difficulty in a comparison of the excited state properties of the literature examples lies in the different levels of structural organisation of the porphyrin and ligand moieties. While the two groups in 1 are restrained in a cofacial arrangement by a flexible strap, the phenyl bridges of 2 provide a linear alignment of the groups and yet allow free rotation about the s-bond linkages. In a further contrast, 3 enforces a rigid planar orientation of the groups, with a high degree of electronic coupling provided by the fused aromatic linkage. The use of low temperature conditions which are required for the photophysical activity of terpyridine complexes of 2[5] presents further practical difficulties in the evaluation of such compounds.
This account reports the use of a Diels-Alder approach based on the use of ligand-functionalised dienes with porphyrin dienophiles to obtain porphyrin-ligand couples. As we will demonstrate, this simple approach allows straightforward access to large, difunctional systems in which the two effector sites are structurally separated in a well defined manner by a fused alicyclic spacer. This strategy provides a new degree of organisation of the porphyrin and ligand groups and presents the opportunity for the systematic evaluation of distance and orientation effects in photophysical studies.
A common synthetic strategy was employed in this study (see Scheme
1) to prepare the selection of porphyrin-ligand couples, each
unique in the structural arrangement of the effector groups.
The starting point for each synthesis was the quinoxaline-fused
diamino-porphyrin 4, first described by Crossley[8].
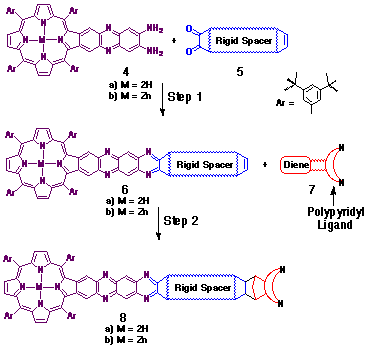
The remaining major steps en route to the target molecules (8)
comprised an a-dione-o-diamine condensation (Step 1) with
a pre-prepared rigid spacer a-dione
unit (5), followed by a stereoselective cycloaddition at
the alkene with a ligand-functionalised diene (7) (Step
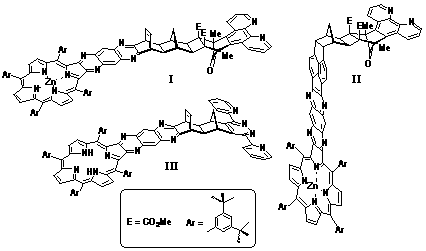
2). Typical products I-III obtained via
this methodology are illustrated in Figure 2.
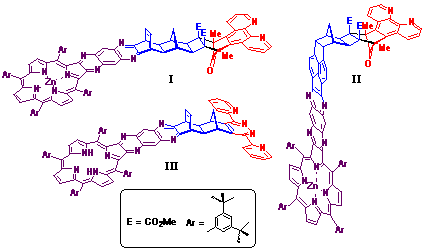
While the bidentate metal complexes of ligand I-III are yet to be prepared, structures I and II were prepared as the metallated porphyrin (Zn2+) species in order the allow the synthesis of the dimetallic [Zn(II)-porphyrin]-[Ru(II)-polypyridyl] structures in future studies. In contrast, structure III was prepared as the porphyrin free base which is valuable for the preparation of various homodimetallic species.
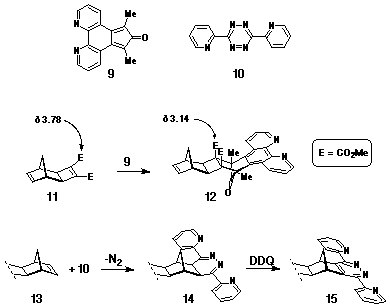
Ligand Cycloaddition Reagents
The ligand reagents used as dienes in Step 2 of the synthesis included 7,8-diazaphencyclone 9 (DAPC)[9] and 3,6-di-(2'-pyridyl)-s-tetrazine 10[10] (Scheme 2), which were selected due to their cycloaddition character and stereoselectivity. The ability of 9 to react as a normal electron-demand diene towards electron-deficient dienophiles, eg cyclobutene-1,2-diester 11, with high stereoselectivity has been well documented[9,11,12]. In the synthesis of model compounds such as 12[12] (Scheme 2), the stereoselectivity has been assigned on the basis of 1H NMR data where the methyl resonance of the activating ester groups is observed to move significantly upfield following cycloaddition. This effect has been attributed to the shielding effect of the phenanthroline ring which, in the pictured stereochemistry, is proximate to the ester groups.
In contrast, the s-tetrazine 10 has been used as an
inverse electron-demand diene with norbornene p-bonds, eg
13, to give the dihydro-pyridazine 14, which can
be oxidised to the fused dipyridyl pyridazine (DPP) ligand 15[13,14].
Synthesis of Porphyrin-Ligand I
The preparation of I involved the synthesis of difunctionalised
spacer 17 containing both an a-dione
and a dienophilic p-centre. The electron-deficient cyclobutene
was formed by adding dimethyl acetylenedicarboxylate (DMAD) to
16[15] via a metal catalysed [2+2] cycloaddition
under Mitsudo conditions[16,17] (Scheme 3) to provide the
dienophilic character necessary for reaction in Step 2 of the
synthesis. This is the first example which shows that a-diones
are stable to the Mitsudo coupling conditions.
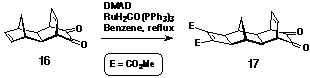
Porphyrin-diamine 4a was condensed with a-dione
17 to give the free base porphyrin 18, which was
metallated with Zn(OAc)2 to provide 19.
Target structure I was obtained following the cycloaddition
of 19 with DAPC (9) (Scheme 4).
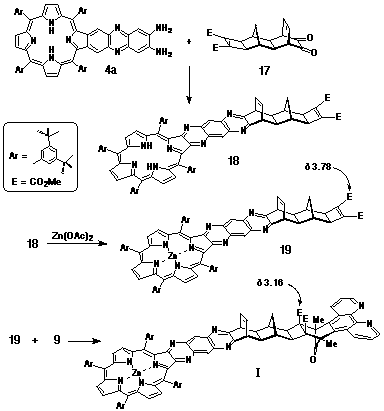
The stereostructure of I was confirmed by the characteristic upfield shift of the methyl ester resonance (d3.16), indicative of the Alder endo-addition of DAPC.
Synthesis of Porphyrin-Ligand II
The bent frame spacer 22, responsible for enforcing the
bent geometry in product II is based upon pyracyloquinone
20, which was first reported by Trost in 1969[18]. Following
Trost's original synthetic procedure, the Diels-Alder adduct 21
was isolated and reacted further with DMAD under standard Mitsudo
conditions to provide the activated cyclobutene derivative 22
(Scheme 5).

The synthesis of porphyrin-ligand II followed an analogous
procedure for the preparation of I. a-Dione
22 was condensed with porphyrin-diamine 4a to give
fused porphyrin 23, which after conversion to its metallated
species 24, was reacted with DAPC (9) to provide porphyrin-ligand
II (Scheme 6).
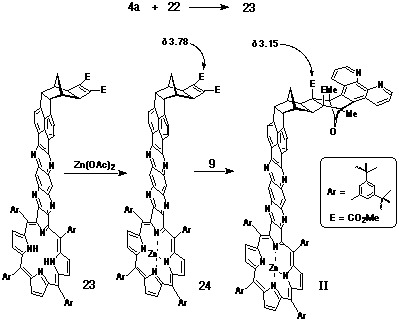
The stereochemistry of II was consistent with previous
DAPC additions to cyclobutenes, with the upfield shift of the
methyl ester groups (d3.15) once again
indicative of the pictured stereochemistry.
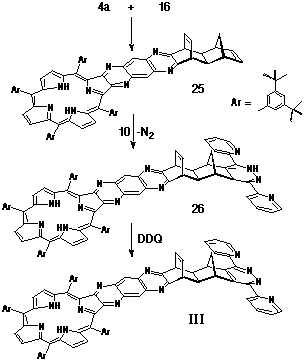
Synthesis of Porphyrin-Ligand III
The preparation of porphyrin-ligand III again employed
the spacer 16 as its key structural unit. Porphyrin-diamine
4a was condensed with a-dione
16 to give structure 25 (Scheme 7) which,
in turn, was reacted with the inverse electron-demand diene 3,6-di-(2'-pyridyl)-s-tetrazine
10. Following the loss of dinitrogen, the resultant dihydropyridazine
26 was oxidised with DDQ to afford the porphyrin-pyridazine
couple III.
Molecular modelling of products I-III was conducted
at the AM1 level of theory to establish the structural relationship
between the porphyrin and ligand groups. Due to the large number
of atoms in each structure, modelling was performed on a truncated
structural system: the t-butyl substituents on the porphyrin
periphery were removed, diester substituents were contracted to
anhydrides, and the porphyrin was modelled as the free base.
The geometrical data describing the mutual orientation of the porphyrin and ligand groups is summarised in Table 1, with the measurements defined by the schematic structure 27.
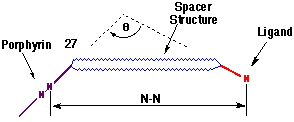
The variants responsible for the individual geometries exhibited by I-III are the structure of the rigid spacer unit and the nature of the ligand. Specifically, the large change in the angular relationship between effectors of I and II can be attributed to the unique structural character of the a-diones 17 and 22, illustrating the importance of such molecular scaffolds in supramolecular chemistry. In addition, the primary difference between I and III is associated with the choice of the ligand terminus which provides variation in both structure and coordination character. The synthesis of I (and II) capitalised upon the use of the electron-deficient cyclobutene dienophile to introduce the phenanthroline ligand via the stereospecific cycloaddition of DAPC (9). In each case, the stereoselectivity of the addition was the same and was easily diagnosed following the upfield shift of the ester methyl resonances (0.7 ppm). This effect has been found to be universal in cycloadditions of DAPC to cyclobutene diesters[9,11,12].
In contrast, the synthesis of III utilised the strained norbornene p-bond in the inverse electron demand cycloaddition with 3,6-di-(2'-pyridyl)-s-tetrazine 10 to afford the fused dipyridyl pyridazine (DPP) ligand. The use of this cycloaddition for the fusion of the DPP ligand to norbornenes has been well documented[13,14]. Although the DPP ligand is capable of forming 1:1 photoactive complexes with the larger d6 metals[20], its ability to form grid-like inorganic architectures[21] due to the formation of 1:2 complexes with smaller metal ions (eg Co2+, Ni2+, Cu+ and Cu2+),[22-25] provides a wider scope for structures such as III in supramolecular chemistry.
The modular approach adopted in this study presents a straightforward method for the synthesis of high molecular weight species in which the spacial relationship between effector groups can be strictly controlled. This control, which provides considerable advances over structures where the ligand unit is free to rotate, is derived from two features: the high level of structural rigidity provided by the spacer molecule; and the stereospecificity observed in the cycloaddition of the ligand reagent. As illustrated in this account, the choice of bridging units and ligand groups can be utilised to provide a high degree of variability between examples. Future work will be directed at the synthesis of metal complex-porphyrin couples and an investigation of their photophysical properties.
Porphyrin-diamine 4[8], 7,8-diazaphencyclone (9)[9], and 3,6-di-(2'-pyridyl)-s-tetrazine (10)[10] were prepared according to the literature procedures.
General Procedure for Metallation of Porphyrins
A solution of the porphyrin free base in CH2Cl2 was heated on a steam bath while a solution of Zn(OAc)2 in methanol was added in several portions. Heating continued (five minutes) until TLC analysis confirmed the absence of the free base. Upon cooling, the solution was washed with water and dried over Na2SO4. The material was purified by column chromatography on silica, eluting with 1:1 CH2Cl2/petroleum spirit.
Synthesis of a-dione 17
Molrac dione 16[15] (540 mg, 2.7 mmol) was dissolved in benzene (40 ml) with excess dimethylacetylene dicarboxylate (1 mL, 1.15 g, 8.1 mmol) and RuH2CO(PPh3)3 (125 mg, ~5 mole %). The solution was heated at 80oC for two days under argon. Removal of the solvent yielded a brown oil, which was washed through a plug of silica with 2% methanol/CHCl3. The eluted material was further purified by column chromatography on silica by elution with CHCl3. Combined fractions were evaporated to dryness to give a yellow oil which crystallised on standing. Yield: 436 mg, 47%. 1H NMR (300MHz, CDCl3) d 1.11 (1H, d, J = 12.1 Hz); 2.07 (2H, s); 2.14 (1H, d, J = 12.1 Hz); 2.47 (2H, s); 2.72 (2H, s); 3.63 (2H, m); 3.78 (6H, s); 6.38 (2H, m).
The dione diester was used without further characterisation.
Synthesis of Porphyrin-DAPC adduct I.
Diamino-porphyrin 4a (86 mg, 72 mmol) and a-dione 17 (35 mg, 102 mmol) were dissolved in deoxygenated pyridine (3 mL) and the solution stirred at room temperature for three days under nitrogen. The solution was evaporated to dryness and the mixture purified by column chromatography on silica, initially eluting with 1:1 CH2Cl2/petroleum spirit followed by CHCl3. The major porphyrinic band (Rf 0.5) was taken to dryness and examined by 1H NMR to confirm the formation of 18. Yield: 98 mg, 90%. The porphyrin was metallated with zinc acetate before being used in the next step.
A solution of the metallated porphyrin 19 (20 mg, 13 mmol) and DAPC (9) (6 mg, 23 mmol) in CHCl3 (400 mL) was heated at 90oC in a sealed tube for 16 hours. The reaction mixture was purified by column chromatography on silica, eluting with CHCl3. The major porphyrinic band (Rf 0.2) was taken to dryness and the product recrystallised from CH2Cl2/petroleum spirit. Yield: 14 mg, 59%. 1H NMR (300 MHz, CDCl3) d 1.52 (m, 73H); 2.01 (s, 6H, 2xCH3); 2.05 (s, 2H); 2.13 (s, 2H); 2.59 (d, J = 11.9 Hz, 1H); 2.91 (s, 2H); 3.16 (s, 6H, 2xCO2CH3); 4.39 (s, 2H); 6.63 (t, J = 3.7 Hz, 2H); 7.66 (m, 2H); 7.80 (d, J = 1.7 Hz, 2H); 7.96 (d, J = 1.7 Hz, 2H); 7.99 (t, J = 1.7 Hz, 2H); 8.01 (d, J = 1.7 Hz, 2H); 8.09 (d, J = 1.7 Hz, 4H); 8.52 (d, J = 8.0 Hz, 2H); 8.61 (s, 2H); 8.87 (s, 2H); 8.98 (s, 4H); 9.20 (d, J = 4 Hz, 2H). HR-MS (electrospray): 1825.5 M+, 912.9 M2+.
Synthesis of a-dione-cyclobutene 22.
a-Dione 21[18] was treated in the same fashion as that described for the preparation of 17. Yield: 69% mp: 276oC (decomp) 1H NMR (300 MHz, CDCl3) d 1.86 (s, 2H); 2.17 (s, 2H); 2.93 (s, 2H); 3.76 (s, 6H); 4.41 (s, 2H); 7.67 (d, J = 7.1 Hz, 2H); 8.06 (d, J = 7.1 Hz, 2H).
Synthesis of Porphyrin-DAPC adduct II.
A solution of diamino-porphyrin 4a (44 mg, 37 mmol) and a-dione 22 (20 mg, 48 mmol) in deoxygenated pyridine (1.5 mL) was stirred at room temperature for two days under nitrogen. The solution was taken to dryness and the residual material purified by column chromatography on silica, eluting with CHCl3. The major porphyrinic band (Rff 0.5) was taken to dryness and examined by 1H NMR to confirm the formation of 23. Yield: 44 mg, 76%. Following metallation of the porphyrin with Zn(OAc)2, a solution of porphyrin-cyclobutene 24 (13 mg, 7.9 mmol) and DAPC (9) (5 mg, 9.6 mmol) in CHCl3 (400 mL) was heated in a sealed tube at 90oC for seven days. The reaction mixture was purified by column chromatography on silica, eluting initially with CH2Cl2 to remove starting material, followed by 5% methanol/CHCl3 to elute the product. Yield: 12 mg, 80%. 1H NMR (400 MHz, CDCl3) d 1.48-1.61 (m, 80H); 2.02 (d, J = 11 Hz, 1H); 2.42 (d, J = 11 Hz, 1H); 3.15 (s, 8H, 2xCO2CH3 + 2H); 4.16 (s, 2H); 7.57 (m, 4H); 7.80 (t, J = 1.7 Hz, 2H); 8.01 (m, 4H); 8.07 (s, 2H); 8.10 (m, 4H); 8.39 (d, J = 8.6 Hz, 2H); 8.46 (d, J = 6.8 Hz, 2H); 8.84 (s, 2H); 8.86 (s, 2H); 8.93 (d, J = 4.6 Hz, 2H); 8.96 (d, J = 4.6 Hz, 2H); 9.11 (d, J = 4 Hz, 2H).
Synthesis of Porphyrin-Tetrazine adduct III.
A solution of porphyrin diamine 4a (46 mg, 38 mmol) and a-dione 16 (12 mg, 56 mmol) in deoxygenated pyridine (1.5 mL) was stirred at room temperature for two days under nitrogen. The solution was taken to dryness and the residue purified by column chromatography on silica, eluting with 1:4 CH2Cl2/petroleum spirit. The major porphyrinic band (Rf 0.3) was taken to dryness and recrystallised from CH2Cl2/petroleum spirit. Yield: 50 mg, 93%. HR-MS (electrospray): 1359.9 (M+). Porphyrin-adduct 25 (45 mg, 33 mmol) was reacted directly with 3,6-di-(2'-pyridyl)-s-tetrazine (10) (11 mg, 44 mmol) by heating at reflux in CHCl3 solution (2 mL) for 30 minutes. DDQ (13 mg, 57 mmol) was added and heating continued for a further four hours. On cooling, the solution was taken to dryness and the residue purified by column chromatography on silica, initially eluting with 1:1 CH2Cl2/petroleum spirit, followed by CHCl3 to elute the product. Yield: 33 mg, 64% 1H NMR (400 MHz, CDCl3) d -2.42 (s, 2H); 1.49-1.53 (m, 72H); 1.70 (d, J = 8 Hz, 1H); 2.49 (s, 2H); 3.23 (d, J = 8 Hz, 1H); 4.55 (t, J = 2.6 Hz, 2H); 4.79 (s, 2H); 6.85 (t, J = 2.6 Hz, 2H); 7.43 (m, 2H); 7.79 (t, J = 1.7 Hz, 2H); 7.90 (m, 2H); 7.94 (s, 2H); 8.00 (s, 2H); 8.07 (d, J = 1.7 Hz, 4H); 8.47 (s, 2H); 8.55 (d, J = 6 Hz, 2H); 8.75 (s, 2H); 8.86 (d, J = 3 Hz, 2H); 8.94 (d, J = 4 Hz, 2H); 8.97 (d, J = 4 Hz, 2H).
Acknowledgments
Deakin University Analytical Service is thanked for conducting electrospray MS on products I-III. ACS thanks Central Queensland University for the award of a postgraduate scholarship, and acknowledges financial assistance from the CMA. This work was funded by the Australian Research Council and Central Queensland University.
(1) Schneider, H.-J., Durr, H. Frontiers in Supramolecular Organic Chemistry and Photochemistry; VCH: Weinheim, 1991.
(2) Balzani, V., Scandola, F. Supramolecular Photochemistry; Ellis Horwood Limited: Avon, 1991.
(3) Juris, A., Barigelletti, S., Campagna, S., Balzani, V., von Zelewky, A. Coord. Chem. Rev. 1988, 84, 85-277.
(4) Wasielewski, M. R. Chem. Rev. 1992, 92, 435-461.
(5) Hamilton, A. D., Rubin, H., Bocarsly, A.B. J. Am. Chem. Soc. 1984, 106, 7255-7257.
(6) Collin, J.-P., Harriman, A., Heitz, V., Odobel, F., Sauvage, J.-P. J. Am. Chem. Soc. 1994, 116, 5679-5690.
(7) Odobel, F., Sauvage, J.-P., Harriman, A. Tetrahedron Lett. 1993, 34, 8113-8116.
(8) Crossley, M. J., Burn, P.L., Langford, S.J., Prasher, J.K. J. Chem. Soc., Chem. Commun. 1995, 1921-1923.
(9) Warrener, R. N., Houghton, M.A., Schultz, A.C., Keene, F.R., Kelso, L.S., Dash, R., Butler, D.N. Chem. Commun. 1996, 1151-1152.
(10) Goodwin, H. A., Lions, F. J. Am. Chem. Soc. 1959, 81, 6415-6422.
(11) Warrener, R. N., Ferreira, A.B.B., Schultz, A.C., Butler, D.N., Keene, F.R., Kelso, L.S. Angew. Chem. Int. Ed. Engl. 1996, 35, 2485-2487.
(12) Warrener, R. N., Schultz, A.C., Houghton, M.A., Butler, D.N. Tetrahedron 1997, 53, 3991-4012.
(13) Warrener, R. N., Elsey, G.M., Sanker, I.V., Butler, D.N., Pekos, P., Kennard, C.H.L. Tetrahedron Lett. 1994, 35, 6745-6748.
(14) Warrener, R. N., Elsey, G.M., Houghton, M.A. J. Chem. Soc., Chem. Commun. 1995, 1417-1418.
(15) Warrener, R. N., Johnston, M.R., Schultz, A.C., Golic, M., Houghton, M.A., Gunter, M.J. Synlett. 1998, Accepted for publication.
(16) Mitsudo, T., Kokuryo, K., Shinsugi, T., Nakagawa, Y., Watanabe, Y., Takegami, Y. J. Org. Chem. 1979, 44, 4492- 4496.
(17) Mitsudo, T., Naruse, H., Kondo, T., Ozaki, Y., Watanabe, Y. Angew. Chem. Int. Ed. Engl. 1994, 33, 580-581.
(18) Trost, B. M. J. Am. Chem. Soc. 1969, 91, 918-923.
(19) Warrener, R. N., Elix, J.A., Wilson, W.S. Aust. J. Chem. 1973, 26, 389-407.
(20) Denti, G., Sabatino, L., De Rosa, G., Bartolotta, A., Di Marco, G., Ricevuto, V., Campagna, S. Inorg. Chem. 1989, 28, 3309-3313.
(21) Youinou, M.-T., Rahmouni, N., Fischer, J., Osborn, J.A. Angew. Chem. Int. Ed. Engl. 1992, 31, 733-735.
(22) Andrew, J. E., Blake, A.B., Fraser, L.R. J. Chem. Soc. Dalton Trans. 1975, 800-805.
(23) Ball, P. W., Blake, A.B. J. Chem. Soc. A 1969, 1415-1422.
(24) Andrew, J. E., Ball, P.W., Blake, A.B. Chem. Commun. 1969, 143.
(25) Ball, P. W., Blake, A.B. J. Chem. Soc. (A)
1969, 1415.
{kind=link}