| Structure | Km | Ki | Inhibitor type | |
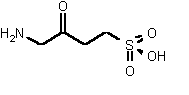 | 69 | 0.26 mM � 0.02 | - | - |
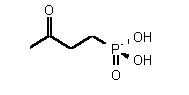 | 70 | 0.37 mM � 0.01 | 25 mM � 3 | competitive |
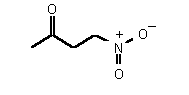 | 71 | 0.26 mM � 0.01 | 18 uM � 3 | competitive |
PBGS is catalysing a complex sequence of events. The kinetics of this process does not reflect one simple fundamental reaction step. Under these circumstances it is clear that the inhibition constants Ki are not the dissociation constant of the inhibitor enzyme complex either. As long as structurally similar inhibitors are used the experimentally determined inhibition constants should still reflect the recognition between the enzyme and the inhibitor.
We synthesised and screened substrate analogues where all positions of the 5-aminolevulinic acid had been modified with the exception of the ketofunction at position 4 (see Figures 35-38).
| Structure | Km | Ki | Inhibitor type | |
| 69 | 0.26 mM � 0.02 | - | - |
| 70 | 0.37 mM � 0.01 | 25 mM � 3 | competitive |
| 71 | 0.26 mM � 0.01 | 18 uM � 3 | competitive |
Figure 35
| Structure | Km | Ki | Inhibitor type | |
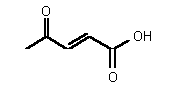 | 72 | 0.35 mM � 0.03 | - | - |
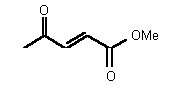 | 73 | 0.25 mM � 0.02 | - | - |
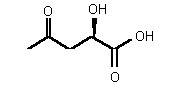 | 74 | 0.40 mM � 0.1 | 0.43 mM � 0.13 | competitive |
Figure 36
| Structure | Km | Ki | Inhibitor type | |
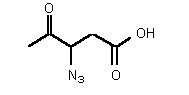 | rac-75 | 0.30 mM � 0.01 | 2.2 mM � 0.4 | competitive |
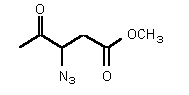 | rac-76 | 0.38 mM � 0.02 | 5.5 mM � 0.9 | competitive |
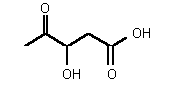 | rac-77 | 0.31 mM � 0.03 | 1.2 mM � 0.4 | competitive |
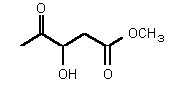 | rac-78 | 0.38 mM � 0.09 | 7.3 mM � 0.6 | competitive |
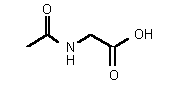 | rac-79 | 0.38 mM � 0.03 | 28 mM � 2 | competitive |
Figure 37
| Structure | Km | Ki | Inhibitor type | |
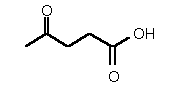 | 80 | 0.36 mM � 0.04 | 1.0 mM � 0.1 | competitive |
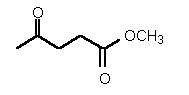 | 39 | 0.35 mM � 0.02 | - | - |
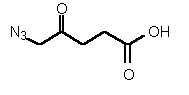 | 81 | 0.20 mM | - | - |
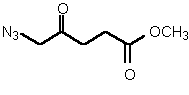 | 82 | 0.35 mM | 3.1 mM | competitive |
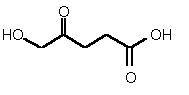 | 83 | 0.40 mM � 0.04 | 0.25 mM � 0.05 | competitive |
 | 84 | 0.36 mM � 0.04 | 2.7 mM � 0.4 | competitive |
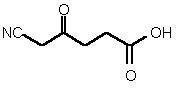 | 85 | 0.30 mM � 0.02 | 60 uM � 15 | competitive |
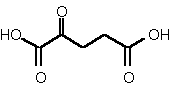 | 86 | 0.37 mM � 0.01 | 19 mM � 2 | competitive |
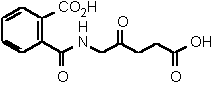 | 87 | 0.28 mM � 0.03 | 27 mM � 4 | competitive |
Figure 38
The most powerful inhibitor was obtained when the carboxylic acid was replaced by the isosteric nitro group 71 (see Figure 35). Changing the substitution at position 2 and 3 gave moderate to good inhibitors. Most of the inhibitors where the substituent at position 5 had been modified were good inhibitors.
The first intermediate postulated by Shemin seemed to us to be an attractive candidate for inhibition studies. The chemical reactivity of the aldol product postulated by Shemin is such that even without the presence of the enzyme pyrrole formation would occur86. Therefore we had to use analogues of the postulated intermediate which lack the amino group involved in the pyrrole formation. The other amino group in the a-position from the ketone had also to be left out, because a-amino ketones dimerise forming dihydropyrazines 284,313. Therefore analogues were synthesised which lack both amino groups (see Figure 39).
| Structure | Km | Ki | Inhibitor type | |
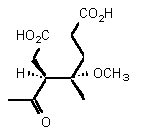 | rac-88 | 0.28 mM � 0.04 | 25 mM � 2 | competitive |
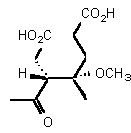 | rac-89 | 0.23 mM � 0.01 | 11 mM � 1 | competitive |
Figure 39
The intermediate postulated by Shemin 163 can exist as four stereoisomers. In order to check if the aldol product is a potential intermediate we needed analogues which are chemically and configurationally stable. To assure that both conditions are fulfilled we decided to study the products rac 88 and rac 89. The two diastereoisomeric pairs of enantiomers rac 88 and rac 89 were studied separately. The inhibition studies showed that one of the pairs of enantiomers rac 88 was a very weak inhibitor, whereas the second pair of enantiomers rac 88 possessed an moderate inhibition constant.
Our method allowed us to synthesise the proposed "mixed pyrrole". The structure could be secured by spectroscopic methods. Repeating Shemin's experiment we could show that indeed a new spot could be detected if 5-aminolevulinic acid and levulinic acid were incubated together with the enzyme PBGS (see Figure 40). However comparing the chromatographic behaviour of the newly formed product with the synthetic material clearly indicated that the two compound are not identical.
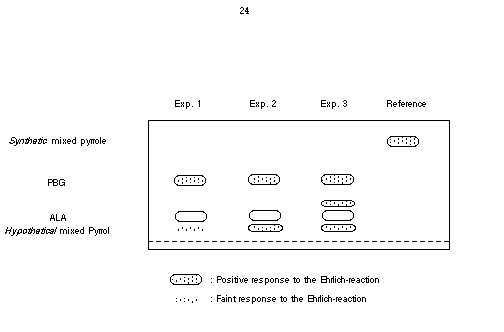
Figure 40 Paper chromatogram of the formation of the mixed pyrrole. Experiment :1 Incubation of PBGS with 5-aminolevulinic acid; Experiment 2: Incubation of PBGS with 5-aminolevulinic acid and levulinic acid; Experiment 3: Incubation of PBGS with 5-aminolevulinic acid and methyl levulinate; Reference: the mixed pyrrole 18 obtained by synthesis
Figure 41
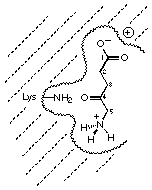
Figure 42 Postulated recognition site of porphobilinogen synthase
The most interesting result is the inhibition studies with the analogues rac 88 and rac 89 of the potential intermediate. Inhibition studies with the racemate of one diastereoisomer rac 88 showed no or only a very weak inhibition (see Figures 42 and 43). The inhibition studies with the racemate of the other diastereoisomer rac 89 gave an inhibition constant of 11 mM.
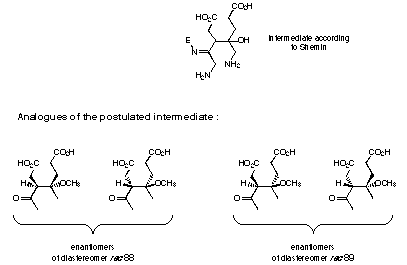
Figure 43 Analogues of the postulated intermediate
The mechanism first postulated by Shemin and latter adjusted by Jordan can now be modified so as to include the newly accumulated knowledge (see Figure 44).
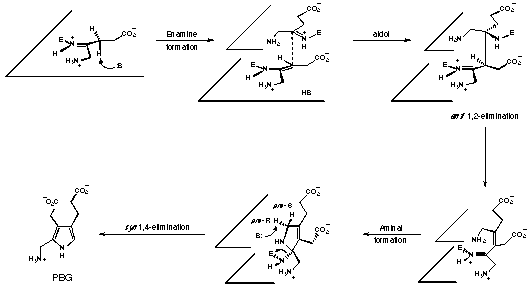
Figure 44 Postulated mechanism for the enzymatic synthesis of porphobilinogen
The relative configuration corresponds to the relative configuration of the better inhibitor. The absolute configuration at the two chiral centres of the aldol intermediate is chosen to be 3R,4S. With this assumption the 1,2-elimination becomes the stereoelectronically preferred trans elimination and the final 1,4-elimination is syn if the pro R hydrogen is eliminated.