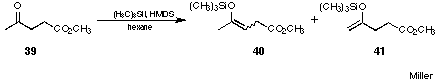
Figure 21
Figure 21
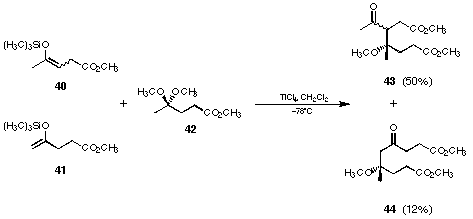
Figure 22 Mukaiyama crossed-aldol reactionCENTER
The b-methoxyketones 43 and 44 so obtained were easier to isolate and the absence of the lactone formation made identification much easier. In a total yield of 62% the two isomeric b-methoxyketone 43 and 44 were obtained by this procedure. The ratio between the two products 43 : 44 ~ 4 : 1 nicely reflected the distribution between the two regioisomeric silyl enol ethers 40 and 41.
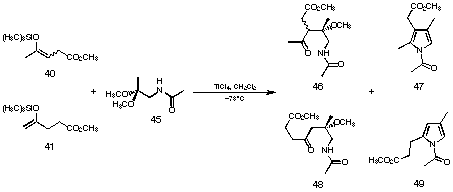
Figure 23 Novel pyrrole synthesis using acetamino acetone (45)
The next step was to apply these reaction conditions to the carbon-carbon bond formation between the silyl enolethers of levulinic acid methyl ester 40 and 41 and the acetal of acetamino acetone (45) (see Figure 23)75. We were able to isolate small quantities of pyrrole 47 already in our first trials. Optimising the reaction conditions we could isolate up to 48% of the pyrrole 47. A large excess of titanium tetrachloride was necessary to obtain good yields of the product. Besides the pyrrolic product 47 we were able to isolate the aldol products 46 and 47 as well. These aldol products 46 and 48 could be cyclised in excellent yield in a separate step using benzene as a solvent and p-toluene sulfonic acid as catalyst.
Trials to use this new pyrrole synthesis for the formation of other pyrroles met with mixed success (see Figure 24)76.
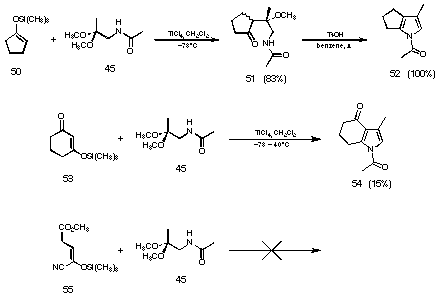
Figure 24 Novel pyrrole synthesis
Treating the silyl enolether of cyclopentanone (50) with the acetal of acetamino acetone (45) gave a mixture of the diastereoisomeric aldol products 51 in 83% yield. These aldol products 51 could be quantitatively transformed into the annelated pyrrole 52 using benzene as solvent and p-toluene sulfonic acid as catalyst. Using deactivated silyl enol ethers like the silylenol ether from 1,3-cyclohexadione (53) the reaction conditions had to be much harsher and still the yield of the pyrrole 54 was disappointingly low. Using the silylenol ether deactivated by the cyano group 55 we were unable to isolate any pyrrolic product.
The difficulties could stem from the fact, that acetamide was used as protecting group. As previously stated by Mukaiyama the crossed aldol reaction does not work if one of the components contains a labile hydrogen atom. Under the influence of titanium chloride hydrogen chloride is formed and thereby the silylenol ether is hydrolysed77.
In order to avoid these problems we changed to a more innocent protecting group, the phthalimido protecting group (see Figure 25).
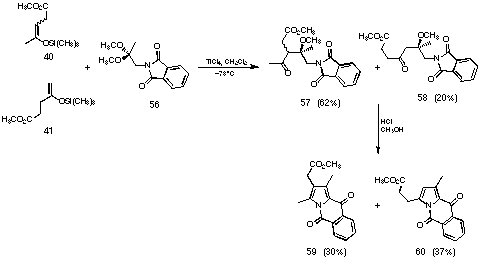
Figure 25 Modified novel pyrrole synthesis
As we had hoped, the yield of the crossed aldol reaction using the dimethyl acetal of phthalimido acetone (56) gave a considerably better yield for the aldol reaction. One of the reasons for the better yield was certainly that the aldol products 57 and 58 are more stable. The hydrolysis of the protecting group proved to be difficult. We finally succeeded in transforming the aldol products 57 and 58 into the pyrrolic compounds 59 and 60. Treating the aldol products 57 and 58 with hydrogen chloride in methanol gave two isomeric tricyclic pyrroles 59 and 60 in moderate yield. The removal of the protecting group was not successful, because the partially hydrolysed intermediate formed the N-acylated pyrrole first and this pyrrole undergoes an intramolecular Friedel-Crafts reaction. These results induced us to search for yet another protecting group.
For the synthesis of the isomerically pure silyl enolether we followed the procedure of Rubottom 78 for the reductive silylation of the corresponding bromoketone. Treating the 3-bromoketone rac 69 with activated zinc in diethylether in the presence of TMEDA and TMS chloride gave only the elimination product (see Figure 26). We could show that the silylenol ether in pure form is stable to extraction with aqueous basic solutions. Therefore the elimination induced by TMEDA must be faster than the heterogeneous reduction with zinc.
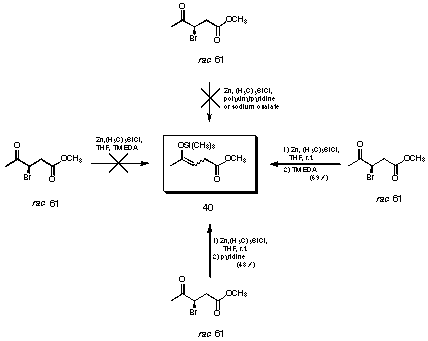
Figure 26 Synthesis of the pure silyl enol ether 40
Using the procedure of Itoh 79,80, silylation with zinc in the absence of a base and quenching the reaction with pyridine, followed by an aqueous work-up allowed to isolate the silylenolether in 49% yield by distillation (see Figure 26). Trying to isolate the silylenol ether avoiding the aqueous work-up, met with no success. Only levulinic acid and polymerised material could be isolated. Trials to use other bases such as polyvinyl pyridine or sodium oxalate did not allow us to remove the zinc salts.
Finally the following procedure allowed to isolate the silylenolether 40 in 69% yield (see Figure 26 and 27). The reductive silylation had to be carried out in the absence of a base. To avoid problems during work-up the zinc salts had to be precipitated using TMEDA to complex the zinc salts and then pentane added. As long as most of the zinc salts could be removed by filtration the silylenol ether 40 could be distilled avoiding the problems of hydrolysis and polymerisation.
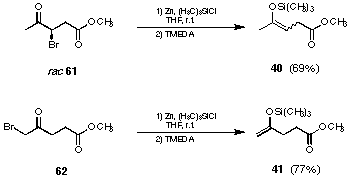
Figure 27 Regioselective synthesis of the silyl enol ethers 40 and 41
To synthesise the regioisomeric silylenolether 41 the 5-bromolevulinic acid methyl ester (62) was treated according to the same procedure and a 77% yield of the silylenol ether 41 could be isolated (see Figure 27).
Instead of the amide protecting group, we decided to use the azido group 273. Already in our first preliminary trials with the mixture of the silyl enol ethers we were able to isolate the aldol products (see Figure 28).
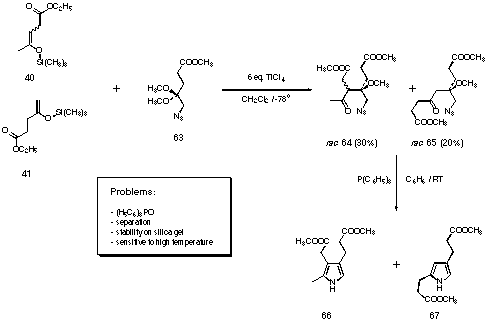
Figure 28 The crossed-aldol reaction followed by the Staudinger reaction applied to the synthesis of the pyrroles 66 and 67
The aldol products were treated with triphenylphosphine in benzene to induce a Staudinger reaction 83-85. We were able to isolate the pyrroles formed 66 and 67. The isolation of the pyrroles was difficult too. The separation of the triphenylphosphineoxide from the alkylpyrroles 66 and 67 was delicate. The separation of the isomeric aldol products rac 64 and rac 65 was also diffucult and the yields were not satisfactory.
The substitution of the amido group by the azido group offered a solution to our problems with the crossed aldol reaction but the overall yield of reaction sequence was still not satisfactory. To use regioisomerically pure silyl enol ethers 40 and 41 considerably improved the yield of the aldol process (see Figure 29).
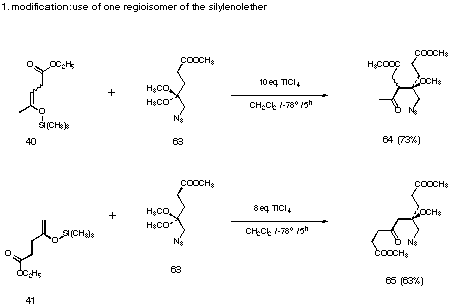
Figure 29 The crossed-aldol reaction using the pure silyl enol ethers 40 and 41
At least 1 equiv. f titanium tetrachloride per complexing functional group was used. This procedure gave in all cases studied the best yields.
In our first trials we used the Staudinger reaction followed by an aza-Wittig reaction to create the pyrrole ring. Two major problems arouse using these reaction conditions (see Figure 28). The isolation of the pyrroles in pure form proved to be delicate. In order to avoid these problems we replaced the triphenylphosphine by triethylphosphine (see Figure 30). The triethylphosphine oxide formed is water soluble and can therefore be removed by extraction.
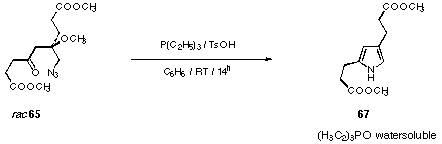
Figure 30 Modified Staudinger reaction
Catalytic reduction is another mild method to transform the azido group into the corresponding amine. Using Pd/C as catalyst and methanol as solvent the aldol products could be reduced (see Figure 31). The amino ketone formed spontaneously the corresponding pyrrole. The work-up using these conditions was extremely easy.
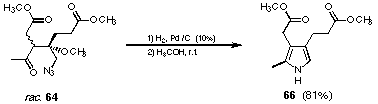
Figure 30 Catalytic reduction
The new two-step pyrrol synthesis allows to synthesise mono-, di-, tri- and tetra-alkylpyrroles in good yield (see Figure 32 and 33). In the case of the cyclopentenyl derivative no pyrrole could be isolated. The yield of pyrrole obtained by reduction was extremely low for the annelated pyrroles. The products of the aldol reaction were mainly the olefins. Probably the a,b-unsaturated olefin is reduced first, which could explain, why no pyrrole was isolated.
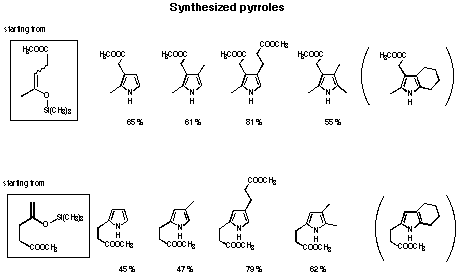
Figure 32
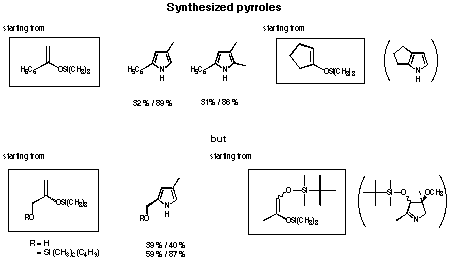
Figure 33
The motivation for the development of this synthesis has been the proposed pathway for the biosynthesis of PBG (9). In view of the results of our model studies we can interpret the postulated mechanism for the enzymatic formation of PBG (9). The foremost task of the enzyme would be to induce the crucial carbon-carbon bond formation (intermediate 68)(see Figure 34).
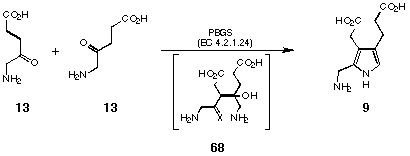
Figure 34 Proposed intermediate of the biosynthesis of porphobilinogen