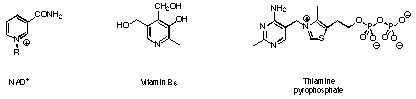
Figure 1 Important coenzymes containing heterocycles.
Figure 1 Important coenzymes containing heterocycles.
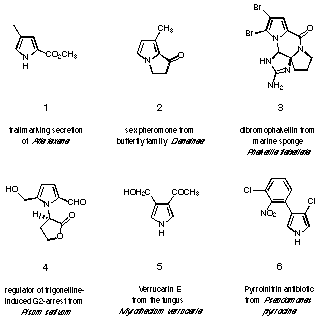
Figure 2 Monopyrrolic natural products.
An important class of pyrrole derived natural products containing more than one pyrrole ring are neotropsin 7 and distamycin 8 which bind to the minor groove of DNA (see Figure 3) 12. They contain a series of pyrrole rings which are linked by amide bonds.
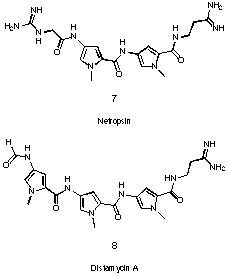
Figure 3 DNA-binders neotropsin 7 and distamycin 8.
Porphobilinogen (= PBG 9) is a remarkable exception to this rule (see Figure 4). PBG 9 is a trisubstituted pyrrole which contains only alkyl substituents.
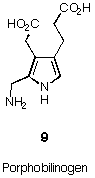
Figure 4 Porphobilinogen
PBG is a dedicated intermediate in the biosynthesis of tetrapyrroles 17-19. The tetrapyrrolic pigments 10 - 12 play an important role for central processes of live (see Figure 5). They are universally distributed and have therefore been named the "pigments of life" 20,21.
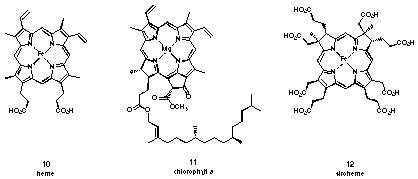
Figure 5 Some "pigments of life"
Five synthetic strategies have been used for the synthesis of PBG 9 (see Figure 6).
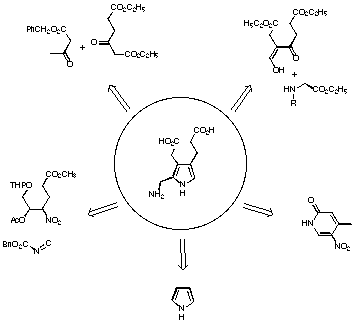
Figure 6 Synthetic strategies for the synthesis of porphobilinogen
In the second strategy developed by Plieninger 39 and Evans 40 the pyrrole ring is formed by condensation of a C3-unit with a C-N-unit. In one case the variant of Kleinspehn of the Knorr synthesis is used, whereas in the second case the ring closure is achieved in a stepwise fashion. In this strategy both the acetic acid and the propionic acid side chains are in place right from the beginning.
The third strategy is due to Frydman and Rapoport 41. They started with a pyridine derivative, which they successfully transformed into a suitably substituted azaindole. Hydrogenation of the azaindole led to the porphobilinogen lactam, which could be hydrolysed to PBG 9.
The fourth strategy stems from Anderson and collaborators, who started with the unsubstituted pyrrole 42,43. Introducing step by step the acetic acid side chain in the b-position, the nitrile group as precursor of the methylamino group in the a-position and the propionic acid side chain in the b'-position finally gave PBG 9.
The fifth strategy was developed by the group of Adamckzyk and was published recently 44. The condensation of an a-acetoxynitro compound with benzyl isocyanoacetate is used to construct the scaffold of PBG.
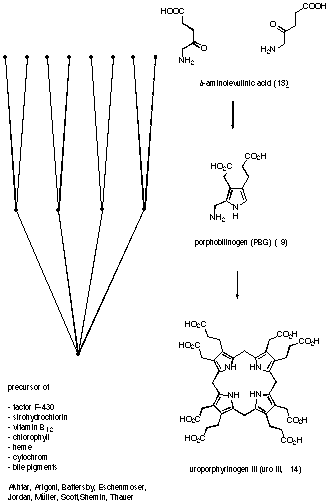
Figure 7 Biosynthesis of uroporphyrinogen III
The first step in the biosynthesis differs between plants and animals (see Figure 7). The final steps of the biosynthesis starting from uroporphyrinogen III (14) vary according to the pigment to be synthesised. In contrast, the central part of the biosynthesis is common to all organisms and for all biosynthetic pathways studied. The physiological importance of the tetrapyrroles and the fact that tetrapyrroles posses a dedicated biosynthetic pathway has stimulated interest in PBG (9).
The two central biosynthetic steps are highly attractive to the synthetic chemist. Using only one molecule as starting material a complex ligand is synthesised in a convergent manner. A mechanistic analysis shows that both processes, the dimeroidization of 5-aminolevulinic acid (13) to PBG and the tetrameroidization of PBG (9) to uroporphyrinogen III (14), belong to the general class of oligomeroidization. Both reactions are exothermic.
From a mechanistic point of view there is one big difference between the two processes. The tetrameroidization can easily be achieved in the absence of an enzyme, whereas it is difficult to realise the dimeroidization in a reaction vessel without enzyme.
Early studies of the reactivity showed that tetrameroidzation of PBG 9 could be achieved easily even without the help of an enzyme (see Figure 8) 55-54.
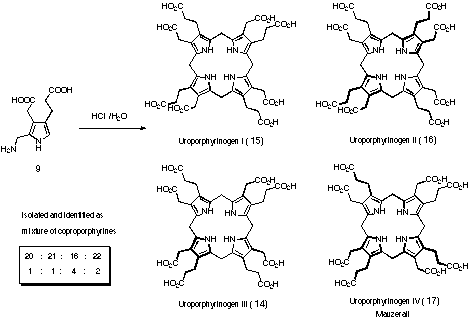
Figure 8 Tetrameroidization of PBG (9)
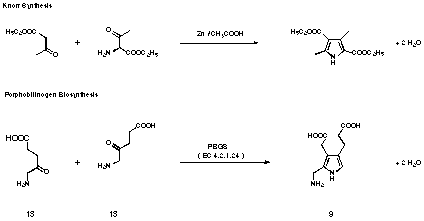
Figure 9 Comparision between Knorr pyrrole synthesis and porphobilinogen biosynthesis
No high resolution X-ray structure of the enzyme PBGS is known so far. Despite the knowledge of almost twenty gene derived proteine sequences for PBGS from different sources, the sequence of events on the enzyme and the mechanistic details of the transformation are stilllargely unknown . The following experimental findings are relevant to the mechanism of PBGS (see Figure 10).
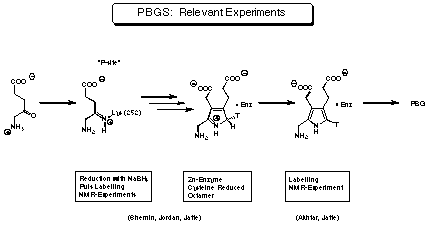
Figure 10 Experiments relevant to the mechanism of PBGS
An argument, which was used in the early studies to postulate a mechanism, was the formation of "mixed pyrroles" 18 and 19 by PBGS from R. spheroides (see Figure 11).
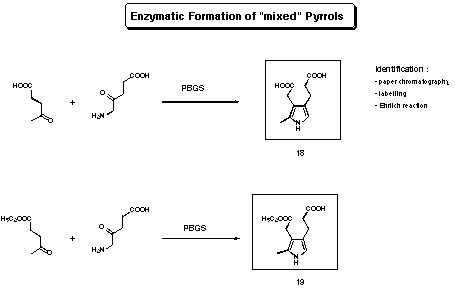
Figure 11 Enzymatic formation of "mixed pyrroles"
Using the enzyme isolated from R. spheroides, Shemin observed the formation of "mixed" or heterologous pyrroles (see Figure 14)55-57. Shemin had only indirect indications for the structures of the two "mixed pyrroles" 18 and 19 (TLC, radioactive labelling). Assuming that the postulated structures of the "mixed pyrroles" are correct, Shemin drew the following conclusion: A pyrrole analogous to porphobilinogen can only be formed between levulinic acid and 5-aminolevulinic acid if the levulinic acid is using the "A-site". If the levulinic acid is bound to the "P-site" the formation of a "mixed" pyrrole is impossible, he therefore deduced, that the sequence of binding is "A-site" first "P-site" second (see Figure 12). Using this postulate and drawing a close analogy between PBGS and class I aldolases, Shemin proposed a mechanism for PBGS.
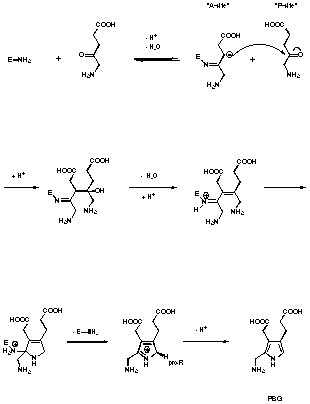
Figure 12 Shemin's mechanism for PBGS
Starting from his observations Jordan postulated an alternative mechanism for the formation of porphobilinogen 58-60 (see Figure 13).
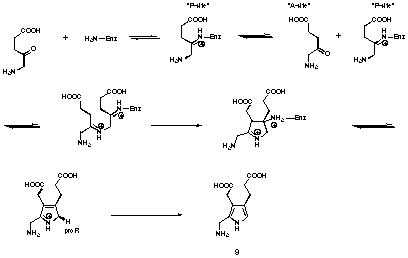
Figure 13 Jordan's mechanism for PBGS
Despite the efforts of several research groups, the mechanism of the enzymatic synthesis of porphobilinogen is not yetestablished . Clear is that the sequence of recognition of the two substrate molecules is "P-site" first, "A-site" second, at least for the bovine liver and the human erythrocyte enzymes. The substrate at the "P-site" forms a Schiff's base to a lysine of the active site. The second substrate may be bound non-covalently to the enzyme. One Zn2+ probably complexed to cysteines helps in the catalytic step. There is good circumstantial evidence, that the enzyme shows half-the-site reactivity. Assuming that the active site is formed at the interface of a dimer would be an attractive interpretation of these observations. Finally, the product forms a relatively stable complex with the enzyme. This observation is in agreement with the fact that the last chemical step, the deprotonation, still occurs at the enzyme.