A few posts back, I contemplated the curly arrows appropriate for the formation of nitrosobenzene dimer from nitrosobenzene,[1] and commented on the odd nature of the N=N double bond formed in this process.[2]. Odd, because the valence bond representation of this dimer (1 below[3]) has two formally positive adjacent nitrogen atoms. An energy decomposition analysis (NEDA[4]) of species 1 showed an unusually small negative interaction energy of -27.6 kcal/mol between the two nitrosobenzene fragments (typical ΔE values ~-130 to -180 kcal/mol[5]), commensurate with the facile equilibrium between two monomers and the dimer[6] A little later I went on to speculate upon a similar theme for the more hypothetical nitric oxide dimer, a species 2 which again has two adjacent +ve charges[7] and even a smaller +ve NEDA for the triple bond! You can imagine discussing these results with organic chemists, who would normally shrink from placing two (formal) positive charges on adjacent atoms.

Browsing (as one does) the CSD crystal structure database, I came across a molecule shown as representation 5 above.[8]. This rang a small alarm bell – why was the central nitrogen atom there shown as neutral? To balance the only +ve charge (on the pyridinium cation), the Ti had a single -ve charge. Representation 3 installs a second +ve charge on the second nitrogen, just as with 1 and 2. The ligand in question (PyN2-1) has an overall charge of -1, and together with the other three negatively charged ligands results in TiIV. The total formal count around the Ti is 6 (from Cp-1) + 2×2 (2Cl-1) + 6 (PyN2-1), making 16e, a fairly normal count for many Ti species and only two short of a filled valence shell of 18e. Alternative representation 4 shows only one +ve and one -ve charge in the molecule, but now the Ti formal valence shell has only 14e.
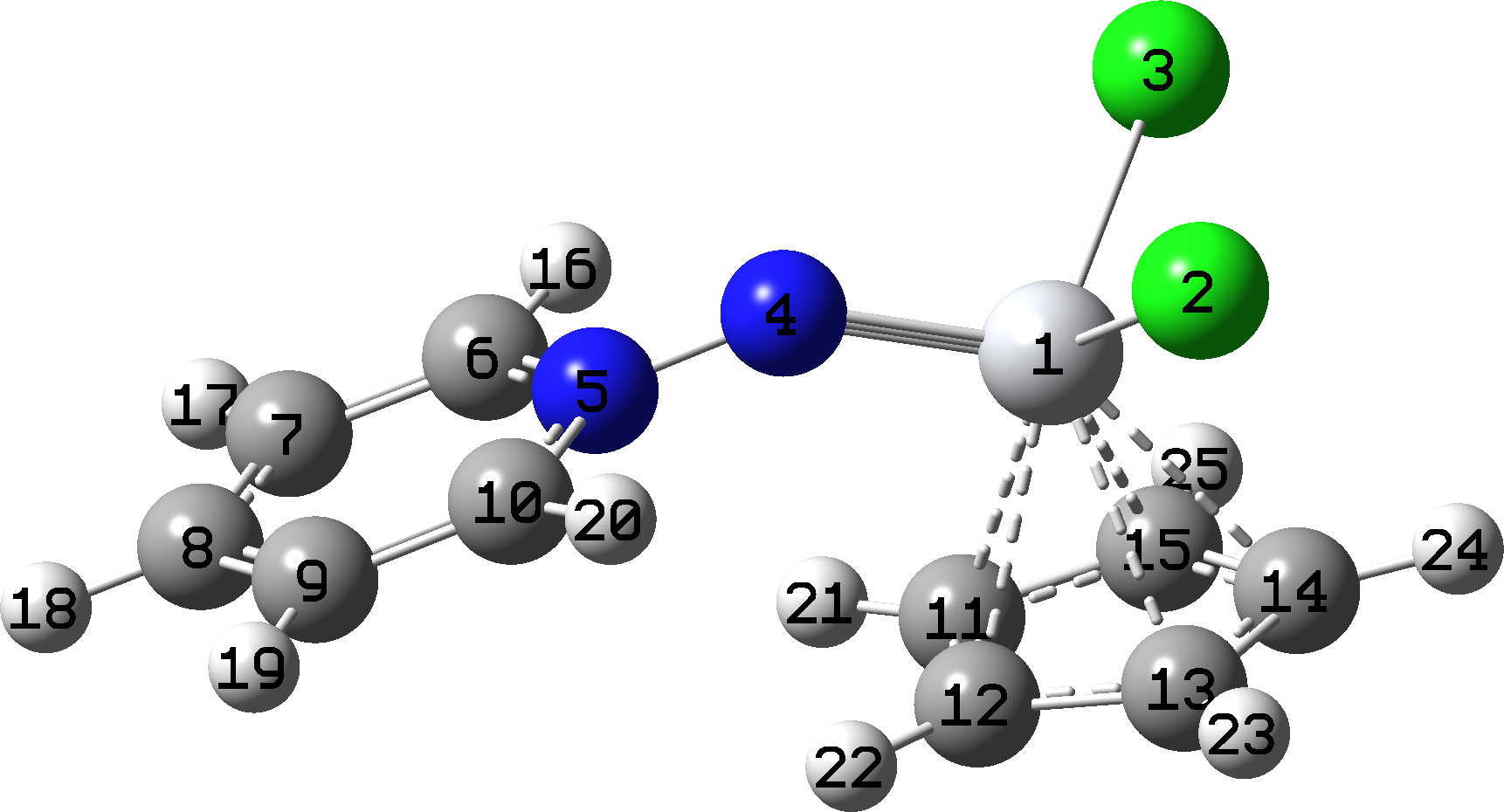
I decided firstly to find out if there was any supporting data for the N≡Ti triple bond as shown in 3 and 5. A search of the CSD database for species with a Ti-N bond (of any order or type) produces the following plot.
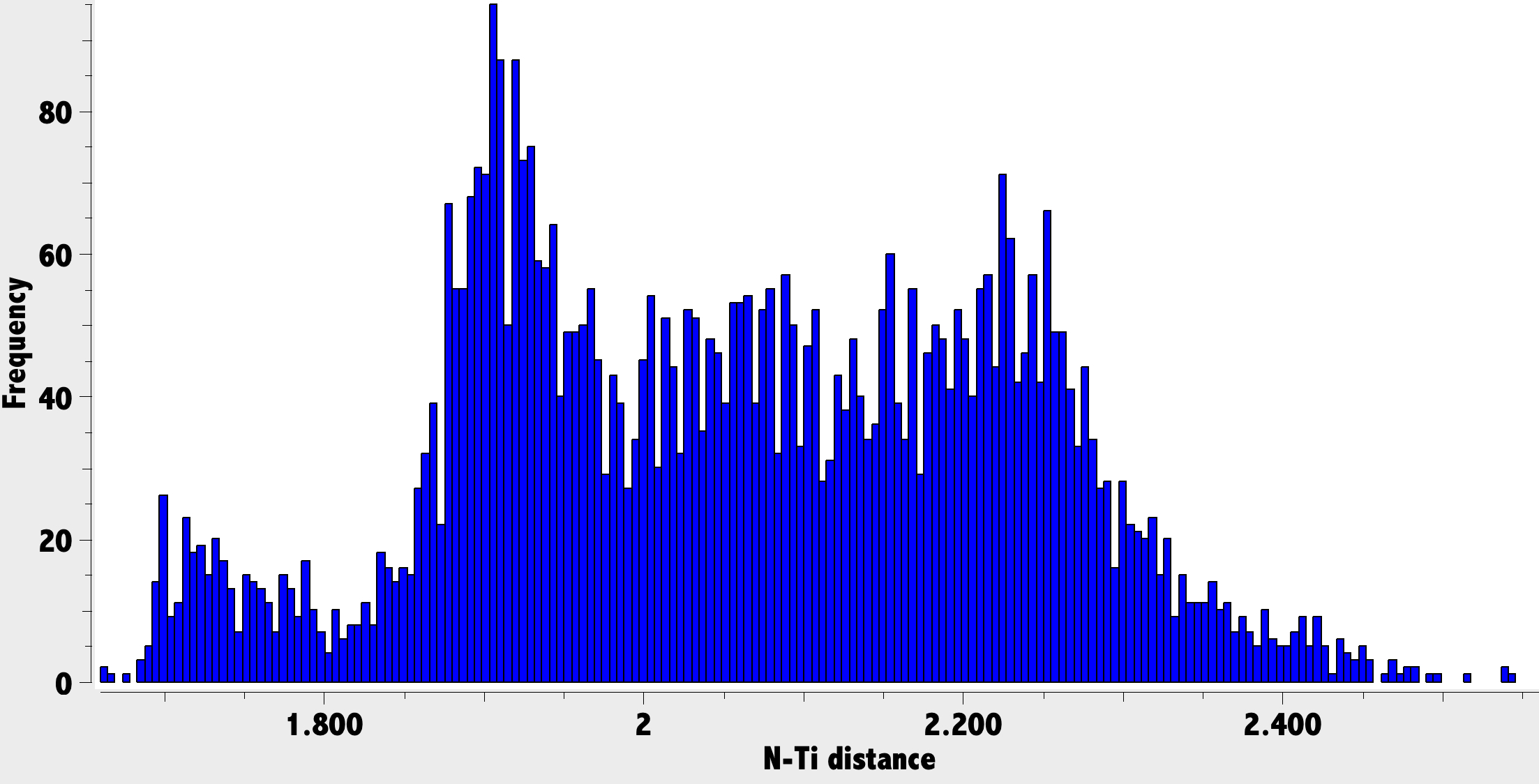
There is a distinct cluster in the region 1.7-1.8Å, which we may assume corresponds to the shortest TiN bonds, presumed to be triple. Two more diffuse clusters are in the region 1.9 – 2.0 (double bonds) and 2.0-2.3Å (single and other bonds). The crystal structure of HOPSUA shows as 1.735Å and hence appears to be in the triple (3) rather than the double bond (4) region. Moreover the measured TiNN angle is 165° whereas 4 might be expected to be more highly bent. The N-N bond length is 1.362Å.
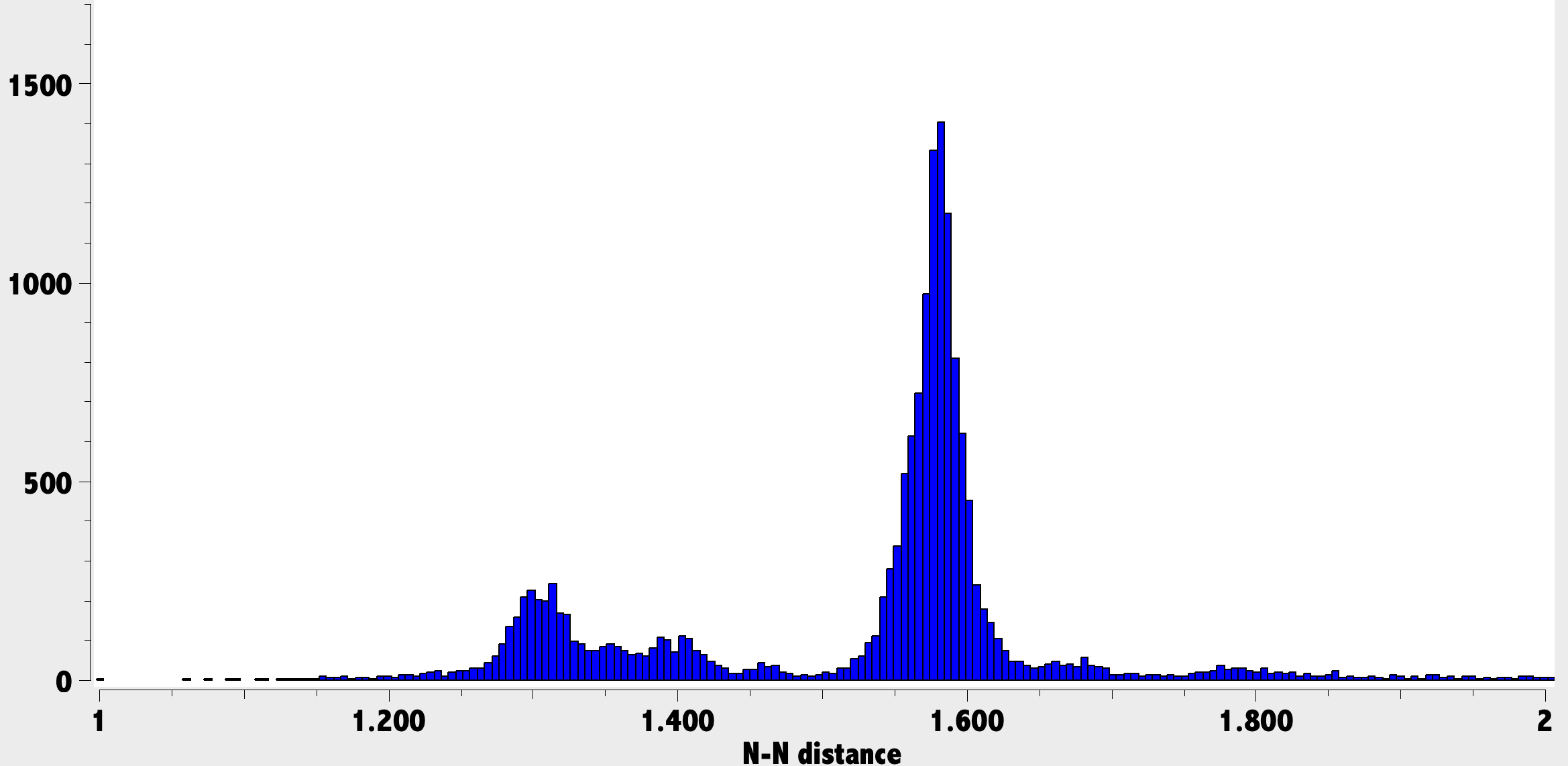
MN15L/Def2-TZVPP and R2-SCAN-3c calculations give respectively TiN 1.784Å/1.806Å and NNTi 147°/152°.[9] The observed NN distance of 1.362Å in HOPSUA and containing an N(+)-N(+) motif (calculated N-N 1.337Å/1.303Å) compares with the distribution below in the CSD, where the main features are ~1.3Å (double), 1.4Å (aromatic) and ~1.58Å (single). So the NN bond order in HOPSUA is rather less than double.
A NEDA (natural energy decomposition analysis)[9] shows that the interaction energy of a neutral singlet pyridine with a singlet NTiCl2Cp fragment (a metal nitrido complex[10]) is -89 kcal/mol. The charge transfer (CT) component is large (-677.4 kcal/mol) because combining two fragments, each deploying a lone pair of electrons to form less than a double bond requires transferring electrons out of this region. This overall interaction energy is larger than for nitrosobenzene, but it is still unusually small for a bond interaction and indeed perhaps a feature of systems which have two (formal) repulsive positive charges on adjacent atoms. For completeness, the interaction energy for the two fragments PyN2 and TiCl2Cp both as doublet states is -83.36 kcal/mol, with a much smaller charge transfer component of -307.4 kcal/mol compared to the NN bond. Despite the apparent disparity between the bond orders of the NN and NTi bonds, they appear to have similar interaction energies! The quartet-quartet decomposition is -184.5, whilst the ionic decompositions [singlet C5H5TiCl2(-) and C5H5N2(+)] vs [singlet C5H5TiCl2(+) and C5H5N2(-)] are respectively -210.6 and -244.6 kcal/mol.
Both 1 and 2 were previously mooted as systems where adjacent atoms bear formal +ve charges, and characterised by their unusually low interaction energies emerging from an energy decomposition analysis. We can now add the known system 3 to this class, albeit its interaction energy being somewhat higher than 1 or 2. There are probably many more of this type out there.
Author
References
- H. Rzepa, "Mechanism of the dimerisation of Nitrosobenzene.", 2025. https://doi.org/10.59350/rzepa.28849
- H. Rzepa, "The mysterious N=N double bond in nitrosobenzene dimer.", 2025. https://doi.org/10.59350/rzepa.29383
- D.A. Dieterich, I.C. Paul, and D.Y. Curtin, "Structural studies on nitrosobenzene and 2-nitrosobenzoic acid. Crystal and molecular structures of cis-azobenzene dioxide and trans-2,2'-dicarboxyazobenzene dioxide", Journal of the American Chemical Society, vol. 96, pp. 6372-6380, 1974. https://doi.org/10.1021/ja00827a021
- C.R. Landis, R.P. Hughes, and F. Weinhold, "Bonding Analysis of TM(cAAC)<sub>2</sub> (TM = Cu, Ag, and Au) and the Importance of Reference State", Organometallics, vol. 34, pp. 3442-3449, 2015. https://doi.org/10.1021/acs.organomet.5b00429
- H. Rzepa, "Energy decomposition analysis of hindered alkenes: Tetra t-butylethene and others.", 2025. https://doi.org/10.59350/rzepa.29410
- K.G. Orrell, V. Šik, and D. Stephenson, "Study of the monomer‐dimer equilibrium of nitrosobenzene using multinuclear one‐ and two‐dimensional NMR techniques", Magnetic Resonance in Chemistry, vol. 25, pp. 1007-1011, 1987. https://doi.org/10.1002/mrc.1260251118
- H. Rzepa, "The even more mysterious N≡N triple bond in a nitric oxide dimer.", 2025. https://doi.org/10.59350/rzepa.29429
- M. Retbøll, Y. Ishii, and M. Hidai, "Synthesis and Reductive N−N Bond Cleavage of Neutral and Cationic Titanium (1-Pyridinio)imido Complexes", Organometallics, vol. 18, pp. 150-155, 1998. https://doi.org/10.1021/om980661t
- H. Rzepa, "Valence bond representations with +ve charges on adjacent atoms? An odd titanium complex analysed.", 2026. https://doi.org/10.14469/hpc/15774
- Carroll, Maria E.., Pinter, Balazs., Carroll, Patrick J.., and Mindiola, Daniel J.., "CCDC 1420931: Experimental Crystal Structure Determination", 2015. https://doi.org/10.5517/cc1jplh9