| [Related articles/posters: 116 106 117 ] |
Introduction. Our group has recently been engaged in the development of a LEGO-type building BLOCK approach to organic synthesis [1], we have investigated the role of a-dione condensation with vicinal diamine as a BLOCK-linking process [2]. In the course of that study we utilised the cycloaddition of dichlorovinylene carbonates with cyclopentadiene and furan as route to the precursors needed for the production of the required norborn-2-en-5,6-dione and 7-oxanorbornen-2-en-5,6-dione respectively. Both cycloaddition reactions have been reported in the literature and structures assigned to the products of reaction by NMR methods, however no details of the NMR method used to make these assignments has been presented. As these assignments rely on chemical shift data alone there was doubt about the reliability of the proposed structures, especially in the cyclopentadiene reaction where only a single adduct was produced. We already had an interest in the use of semi-empirical methods for the assessment of Diels-Alder cycloaddition stereoselectivities [3], so we were curious to apply them to the DCV cycloaditions. First, we set out to clarify the structural assignments by X-ray methods and with these results in hand, to investigate the stereoselectivities of the reaction using more advanced computational methods. In practice, we were able to confirm the original NMR assignments and the computational methods were able to correctly account for the observed stereoselectivities.
We have extended our computational work to include vinylene carbonate as well as dichlorovinylene carbonate as dienophiles and also included fulvene to join with cyclopentadiene and furan in the set of cyclic dienes. In this paper we report ab initio calculations as well as the X-ray data for the key dichlorovinylene adducts 4 and 7.
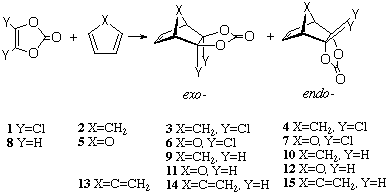
Scheme 1. Cycloaddition reactions modelled in this study
Computational methodology. Initial geometries for our ab
initio studies were generated using the AM1
[4] method using the SPARTAN suite of programs [5]
on Silicon Graphics R5000 or R10000 workstations. Geometry optimizations
were carried out without imposing symmetry or other structural
restrictions. All calculations were performed at the restricted
Hartree-Fock level [6]
with 3-21G and 6-31G* basis sets [7].
Each transition structure was located using a standard routine
within SPARTAN and verified by the possession of only one imaginary
frequency of vibration. The activation energies were also determined
from 6-31G* and MP2(fc)/6-31G* single point calculations on the
RHF/3-21G optimized geometries.
Experimental part. Although the stereochemistry of adducts 4 and 7 were deduced by previous workers on the basis of the NMR data, it is not as reliable in the case of dichlorovinylene carbonate adducts since coupling data is not available. To confirm the stereochemistry of adducts 4 and 7 we have obtained X-ray structures.
CPD and furan DCVC adducts were prepared by methods described by Scharf and Blakenspoor [8, 9]. The crystals for X-ray analysis were prepared by recrystallisation from cyclohexane (compound 4) and diethyl ether (compound 7). For more details see Appendix.
The choice of theoretical method. The objective of this
study was to be able to find method able to predict the stereochemical
outcomes of the cycloaddition reactions rather than their absolute
activation energies. Our previous experience in transition state
modeling Diels-Alder reactions [10, 11]
and literature precident [12, 13] confirm
that relatively low levels of theory give qualitatively correct
result. Consequently we compared single point 6-31G* and MP2/6-31G*
energies obtained from the 3-21G optimized structures to predict
stereochemical outcomes.
Results and discussion. For the qualitative correlation
of reactivity of cyclopentadiene, furan and fulvene in reaction
with DCVC and VC, frontier orbital energy gaps between the reactants
is used. It is assumed that in using the frontier molecular orbital
(FMO) [14] approach, the most reactive
reactant pair will be the one that has the smallest energy gap
between the frontier orbitals (Table 1). Using this criterion
the most reactive pair of addends in this study would be CPD +
DCVC (DE= 12.110 eV). All reactions are predicted to be HOMO diene
controlled and operate as a normal demand Diels-Alder reaction
[15]. The reaction of fulvene with
VC is an exception, as FMO analysis predicts inverse-electron
demand control. These conclusions are supported by the calculations
of electron shift from the diene to the dienophile in the TS (Table
4). FMO theory neglects the steric and electronic interactions
that occur during the course of the reaction. To determine which
TS will have normal or inverse electron demand charge transfer
(qCT) from diene to dienophile was assesed by and Mulliken
[16] and NPA [17]
population analysis (Table 2). In all the transition structures
located, there is only a small qCT from the diene to the dienophile,
suggesting normal electron demand. The largest qCT are found in
the chloro cases and this is smaller for hydrogen owing to the
electron with drawing nature of the chlorine atoms.
Table 1. 6-31G* a-Eigenvalues (eV) for LUMO and HOMO of
investigated speciesa

aDE1=EHOMO(diene)-ELUMO(1), DE2=ELUMO(diene)-EHOMO(1);
DE3=EHOMO(diene)-ELUMO(8), DE4=ELUMO(diene)-E=EHOMO(8);
Table 2. Transition states parametersa
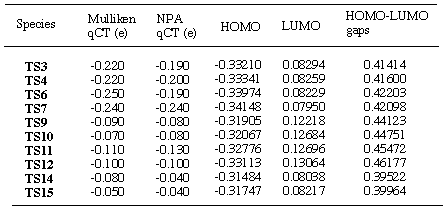
aRHF/6-31G* values; quantum of
charge transfer= qCT
As the reactants draw closer together, the steric and electronic interactions between them become very important for determining the interaction energies of a reactant pair system. One of the best methods for evaluating the reactivity of a species is to estimate the energy of the TS for the corresponding reaction. The total energies of reactants, products and TS are collected in Tables 3 and 4.
Table 3. Total energies (au) of reactants and productsa
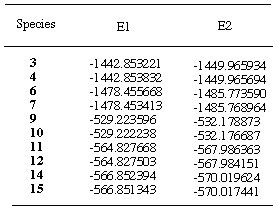
aE1=E(RHF/3-21G); E2=E(RHF/6-31G*//RHF3-21G);
Table 4. Total energies (au) of reactants and transition
statesa
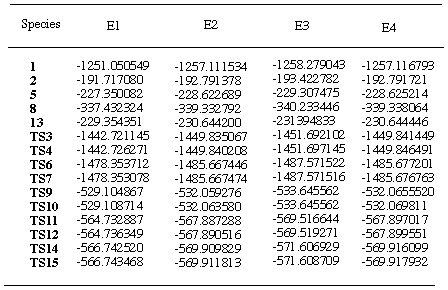
aE1=E(RHF/3-21G); E2=E(RHF/6-31G*//RHF3-21G);
E3=E(MP2(fc)/6-31G*//RHF3-21G); E4=E(RHF6-31G*
Transition state structures. All the transition structures (TS) located support a concerted synchronous mechanism for the cycloaddition reaction (Figures 1-5). Thus, both newly formed C-C bonds have the same bond distance, although these bond lengths differ from case to case. All partially formed or broken C...C single bond lenghts are in the range calculated for hydrocarbon cycloadditions [18]. The shortest C...C bond is found in the furan reactions (2.152 Å) and the longest with CPD (2.267 Å).
The largest geometry changes on going from adducts to TS take place in the dihedral and bond angles. As expected, the amount of geometrical change at the TS is much larger in the diene moiety. For example, the out-of-plane angles in the diene fragments for CPD addition (TS3, TS4, TS9 and TS10) are in the very narrow range of 17.1 - 18.8 o, regardless of the mode of addition. On the other hand, smaller out-of-plane angles are found in the furan and fulvene TS structures (TS6, TS7, TS11, TS12, TS14 and TS15) where the range is 14.5 - 16.5 o.
Furthermore, in all TS structures, the diene olefinic hydrogens are bent out-of-plane towards the dienophile, by a much larger extent than is predicted for products (the C1C6C5H6 dihedral angles are within a range of 7.2 - 8.0 o). This has been observed previously in cyclopentadiene and furan cycloadditions with norbornenes [19]. An explanation for this effect has been proposed by Houk et al. who have assumed that pyramidalization occurs in such a way that p orbitals on C5 and C6 are aligned to maximize orbital overlap with the forming bonds [20, 21].
The frontier molecular orbitals [14]
of the TS structures (Table 4) also indicate that there are additional
stabilisation interactions which decrease the frontier orbital
energies in the favoured isomer (for instance endo- TS
structure 4 in the CPD + DCVC reaction) and therefore make
endo- TS lower in energy.
Control of reaction. Heats of formation of products have been estimated at various levels of theory (Table 3) and show that the exo- products are the thermodynamically more stable. For instance, the difference between products 6 and 7 is estimated to be 12.1 kJ/mol at the 6-31G*//3-21G level. It is obvious that reactions in this study are under kinetic control, otherwise, only endo- products would be experimentally isolated [22].
As expected for Diels - Alder reactions, heats of reaction estimated
at various levels of theory are always very exothermic. For instance,
heats of reaction of 1 and 2 to give cycloadducts
3 and 4 are -165.5 and -164.8 kJ/mol, respectively
at the 6-31G*//3-21G level.
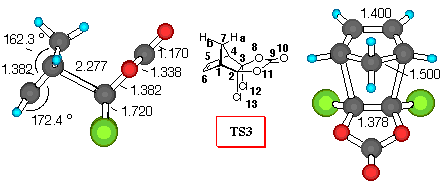
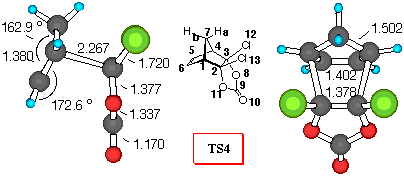
Figure 1. 6-31G* Structures of TS3 and TS4 and numbering
scheme
CPD+DCVC (Figure 1). Dichlorovinylene carbonate (1) reacts with cyclopentadiene yielding the endo- adduct 4 exclusively [8]. As can be seen from inspection of Table 5, all of the theoretical levels employed correctly predict a large preference for the endo- diene approach. Adduct 4 is the preferred one, and relative activation barriers have similar values, regardless of level of calculations (around 13 kJ/mol).
It is also noteworthy that as in many other reactions HF ab initio methods give very high reaction barriers [18]. If the 176 kJ/mol (6-31G* value, formation of product 3) is the correct activation barrier for the cycloaddition reaction, then the reaction would be unlikely to be carried out experimentally. On the other hand, MP2/6-31G*//3-21G* computed activation barriers are too low (6.1 kcal/mol). This would suggest that the reaction should actually be diffusion controlled. Based on the information from the experimental procedure, this is obviously not the case. For calculating highly reliable activation barriers, MP2 or CASSCF methods are required, but for the larger size systems, DFT methods are the most practical approach.
Mulliken population analysis, which gives a qualitative indicator for the amount of electron density shared by two atoms, provides some evidence for secondary orbital interaction between the two reactants, to rationalize observed specificities. It was employed successfully by Houk et al. to explain stereoselectivities in Diels - Alder reactions [23]. In TS3 the H7aC9 (RHF/6-31G* values) overlap density has a negative value of -0.004, indicating a repulsive interaction betwen the carbonyl p-system and the methylene bridge hydrogen. The H7aO8 is much smaller, and the H7aO10 interaction is negligible. Furthermore, the C5Cl12 electron density value of -0.007 also indicates negative secondary orbital interaction. Furthermore, the C7O8 overlap density has a negative value of -0.001, indicating a repulsive interaction betwen the carbonyl oxygen lone pair and methylene bridge carbon. The C7C9 density is much smaller. All these interactions destabilize the exo- mode of approach of cyclopentadiene. In TS4 where DCVC approaches cyclopentadiene in the endo- fashion, the H7aCl12 and C7Cl12 overlap populations are attractive with values of 0.002 and 0.003, respectively. Furthermore, the C5O8 overlap density has a negative value of -0.006, indicating a repulsive interaction (p - n), while the stabilisation of the endo- structure can be explained by the C5C9 and C5O10 stabilizing secondary orbital interactions between diene and carbonyl p- system.
These non-bonding interactions can also be estimated by computing the bond orders. There is no substantial difference between the computed bond orders for the bonds involved in formation of the two isomeric TS structures (C1C2 and C3C4). However, there is a noticeable difference in the secondary orbital interactions between the chlorine atoms of the DCVC moiety with the p- orbitals of the CPD moiety in the TS structures. This C5Cl12 interaction is higher (0.074) in the exo structure than the H7aCl12 secondary molecular orbital overlap in the endo- TS structure (0.016). Furthermore, the H7aC9 secondary molecular orbital overlap (0.039) in TS4 is higher than the corresponding C5C9 secondary molecular orbital overlap (0.023) in TS3, suggesting larger repulsive interactions between the carbonyl system and the methylene bridge in TS3. This properly suggests that the endo- TS structure should have a substantially lower energy than the exo- TS.
The frontier molecular orbitals of the TS structures (Table 2)
also indicate that there are additional stabilisation interactions
which decrease the frontier orbital energies in the TS4
and therefore makes TS4 lower in energy.
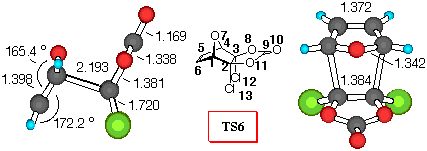
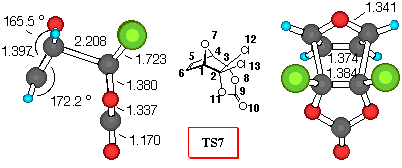
Figure 2. 6-31G* Structures of TS6 and TS7 and numbering
scheme
Furan+DCVC (Figure 2). Dichlorovinylene carbonate (1) reacts with furan yielding a mixture of exo- isomer 6 and endo- isomer 7 in a 2:1 ratio [9]. Inspection of Table 5 shows that all the theoretical levels employed correctly predict the small preference for exo- approach to furan. Adduct 6 is the preferred one, and relative activation barriers, estimated at different theoretical levels have values smaller than 1.6 kJ/mol. The smallest relative energy was estimated by MP2/6-31G*//3-21G method, suggesting that an equal mixture of exo/endo products will be formed.
Recently, it was shown that density functional methods give a very good estimation for activation energies [13, 24]. However, BLYP/6-31G* and B3LYP/6-31G* single point energy estimations on 6-31G* TS structures gave relative activation energies of 0.908 and 0.860 kJ/mol, respectively, favouring exo- TS, which is essentialy the same qualitative result as obtained by HF or MP2 calculations.
If we employ the same analysis of secondary orbital interactions, as in the DCVC+CPD case, we can find that the dominant orbital interactions in TS6 are O7C9, with a positive value for the overlap density of 0.04 (carbonyl p- system and oxygen lone pair atraction). Also, the O7O8 overlap density has a value of 0.002, indicating attractive lone pair - lone pair interactions. Similarly in TS4, the C5O8 overlap density of TS7 has a negative value of -0.004, indicating a repulsive interactions (p - n), while the stabilisation of the endo- structure can be deduced from the C5C9 and C5O10 stabilizing secondary orbital interactions between p- orbital of diene and carbonyl p- system.
Bond order analysis gives the same result - large interactions between carbonyl group and oxygen bridge in TS6, as well as important lone pair - lone pair interactions, and stabilising C5C9 and C5O10 secondary orbital interactions in TS7.
The frontier molecular orbitals of the TS structures (Table 2)
also indicate that there are additional stabilisation interactions
which decrease the frontier orbital energies in the TS7
and therefore make TS6 lower in energy.
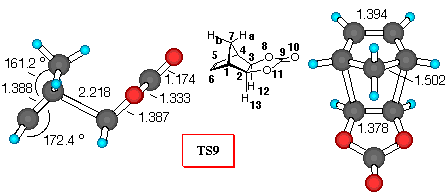
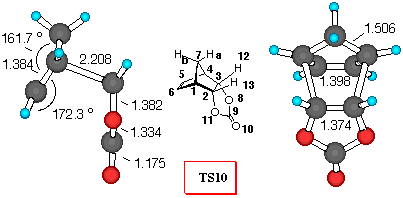
Figure 3. 6-31G* Structures of TS9 and TS10 and
numbering scheme
CPD+VC (Figure 3). Similarly to the DCVC reaction, vinylene carbonate (8) reacts with cyclopentadiene yielding the endo- adduct 10 exclusively [25]. Inspection of Table 5 reveals that all theoretical levels employed correctly predict a large preference for the endo- diene approach. Adduct 10 is the preferred one, and relative activation barriers have similar values, regardless of level of calculations (about 11 kJ/mol), which is 2 kJ/mol smaller than the corresponding DCVC reaction.
If we employ the same analysis of secondary orbital interactions, as in the previous cases, we can find that the dominant repulsive orbital interactions in TS9 are between H7aC9, with a negative value for the overlap density of -0.004 (carbonyl p- system and methylene bridge hydrogen repulsion). In TS10, where VC approaches the cyclopentadiene in an endo- fashion, the H7aH12 overlap population is almost negligible with value of 0.0004, when compared to H7aCl12 value in TS4. Furthermore, the C5O8 overlap density has a negative value of -0.003, indicating an repulsive interactions (p- n), while the stabilisation of endo- structure can be assessed from the C5C9 and C5O10 stabilizing secondary orbital interactions between the p- orbital of diene and the carbonyl p- system.
Bond order analysis leads to the same conclusion - large interactions between the carbonyl group and methylene bridge in TS9, and stabilising C5C9 and C5O10 secondary orbital interactions in TS10.
The frontier molecular orbitals of the TS structures (Table 2)
also indicate that there are additional stabilisation interactions
which decrease the frontier orbital energies in the TS9
and therefore make TS10 lower in energy.
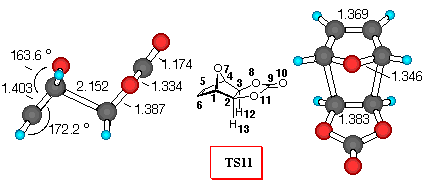
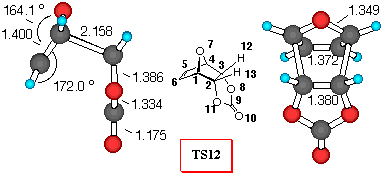
Figure 4. 6-31G* Structures of TS11 and TS12 and
numbering scheme
Furan+VC (Figure 4). Similarly to the reaction with DCVC, vinylene carbonate (8) reacts with furan yielding a mixture of exo- isomer 11 and endo- isomer 12 in 1:4.6 ratio [26]. Inspection of Table 5 reveals that all the theoretical levels employed correctly predict the small preference for endo- approach to furan. Adduct 12 is the preferred one, and relative activation barriers at the 3-21G and 6-31G*//3-21G levels are larger (9.0 and 8.5 kJ/mol, respectively), than those predicted by 6-31G* and MP2/6-31G*//3-21G level of calculations (6.9 and 6.7 kJ/mol, respectively). The smallest relative energy was estimated by MP2/6-31G*//3-21G method, suggesting that the reaction mixture should contain more endo- product.
In this reaction, we can find that dominant orbital interactions in TS9 are O7C9, with a positive value for the overlap density of 0.05 (carbonyl p- system and oxygen lone pair attractions). Also the O7O8 toverlap density has a value of 0.003, indicating attractive lone pair - lone pair interactions. Similarly to the TS7, the C5O8 overlap density of TS12 has a negative value of -0.003, indicating repulsive interactions (p - n), while the stabilisation of endo- structure can be deduced, as in the previously discussed endo- adducts from the C5C9 and C5O10 stabilizing secondary orbital interactions between p- orbital of diene and carbonyl p- system.
The frontier molecular orbitals of the TS structures (Table 5) also indicate that there are additional stabilisation interactions which decrease the frontier orbital energies in the TS12 and therefore make TS12 lower in energy.
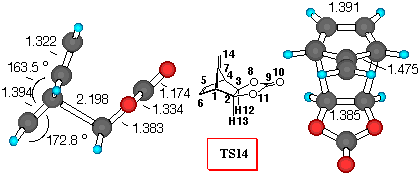
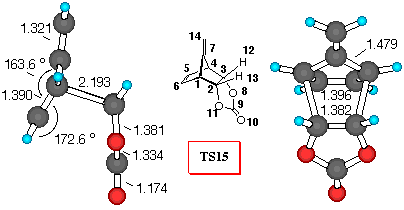
Figure 5. 6-31G* Structures of TS14 and TS15 and
numbering scheme
Fulvene+VC (Figure 5). Vinylene carbonate (8) reacts with dimethyl fulvene yielding a mixture of exo- isomer 14 and exo- isomer 15 in 2:3 ratio [27]. We have chosen this reaction to show the importance of the substituent at the 7- position on the stereochemical outcome of Diels-Alder reactions. For the sake of computational ease, fulvene rather than dimethyl fulvene was investigated.
Inspection of Table 5 indicates that all the theoretical levels employed correctly predict the small preference for endo- approach to fulvene. Endo- adduct 15 is the preferred one, and relative activation barriers are similar at all theoretical levels employed, except 3-21G level, which gives much smaller difference.
In this reaction, we find that the dominant orbital interactions in TS14 are between C7C9, with a positive value for the overlap density of 0.05 (p- p attractions). Also C7O8 the overlap density has a negative value of 0.002, indicating repulsive p- lone pair interactions, which may destabilise the exo- TS. As in all the other cases examined, the C5O8 overlap density of TS15 has a negative value of -0.003, indicating repulsive interactions (p - n), while the stabilisation of endo- structure can be deduced, from the C5C9 stabilizing secondary orbital interactions between the p- orbital of diene and the carbonyl p- system.
The frontier molecular orbitals of the TS structures (Table 5) correctly indicate that there are additional stabilisation interactions which decrease the frontier orbital energies in the TS15 making it lower in energy.
Table 5. Activation and relative energies estimated at
various levels of theory (kJ/mol)a
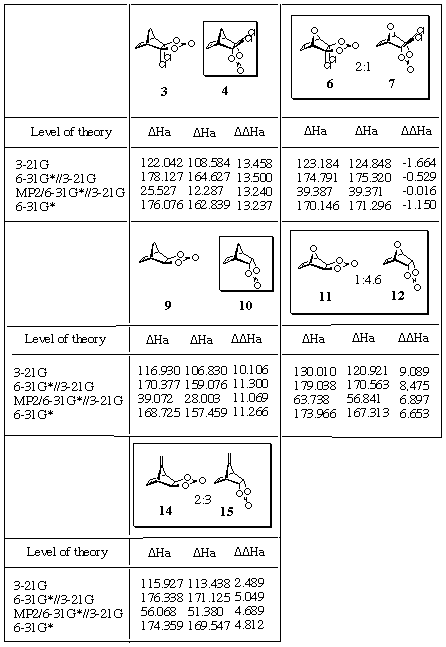
aDDHa=DHa(exo- ) - DHa(endo- );
negative values indicate preference of exo- isomer.
Experimentally obtained products are boxed, alongside the ratio of isomers.
Concluding remarks. The present results demonstrate the ability of ab initio calculations to accurately predict relative reactivities and endo/exo- selectivities for normal electron-demand Diels - Alder reactions in alicyclic systems with cyclic 1,3-dienes. As shown for the reactions of DCVC and cyclopentadiene, as well as DCVC and furan, these reactions undergo kinetic control. The exo/endo- selectivity outcomes exhibited in cycloadditions with DCVC and VC are readily predicted using RHF/3-21G or higher ab initio levels.
The stereochemical preferences were also rationalized by means
of the secondary orbital interactions in the TS. The preference
for endo- over exo- TS in the reaction of CPD with
DCVC or VC can be explained with secondary orbital interactions
between the DCVC and CPD moieties which are present in endo-
but not in the exo- TS structure. The most intense of these
interactions are between methylene hydrogen of CPD and p- carbonyl
orbital of DCVC and VC. In the case of furan, there are smaller
repulsive interactions between the lone pair of oxygen and the
p- carbonyl orbital of DCVC and VC in the exo- TS structures.
Furthermore, lone pair - lone pair interactions make some stabilising
contribution, which may contribute to the formation of both adducts.
Finally, the introduction of an additional p- system in the diene,
as in the case of fulvene, also lead to the formation of two products.
Appendix. The X-ray structures of adducts 4 and 7 and comparison
with calculations.
It is well documented that ab initio calculations predict
geometries very accurately. Indeed, the overall match of RHF/6-31G*
optimised structures of 4 and 7 (Tables 6 and 7)
and experimentally determined geometries is quite good (Figure
6). Although bond lengths and angles are predicted accurately,
this method does not correctly predict the double bond nonplanarity
in norbornenes. The inclusion of electron correlation is essential
to achieve better match with experiment [28].
MP2 calculations or DFT methods (especially B3LYP and B3PW91 functionals)
[29, 30] better represent this pyramidalisation.
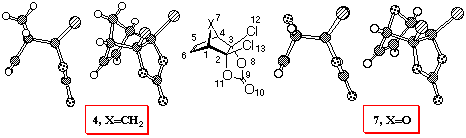
Figure 6. X-ray structures of adducts 4 and 7
Table 6. Comparison of selected structural parameters of adduct
4 calculated with the
6-31G* basis seta
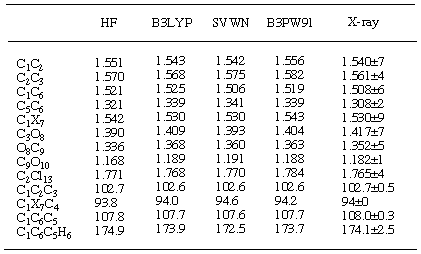
aDistances are given in Ångstroms and angles in degrees. Numbering scheme is
shown at Figure 6.
Table 7. Comparison of selected structural parameters of adduct
7 calculated with the
6-31G* basis seta
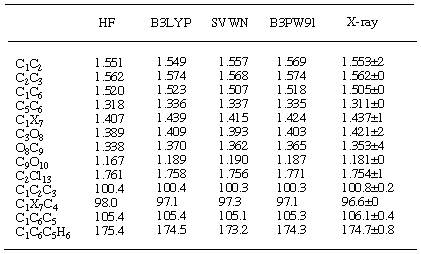
aDistances are given in Ångstroms and angles in degrees. Numbering scheme is
shown at Figure 6.
Acknowledgements. The Australian Research Council is thanked
for funding. We are grateful to Professor Richard V. Williams
for discussions and suggestions during the preparation of this
paper.
References.
[1] Warrener, R. N.; Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A. Chem.Commun. 1997, 1023.
[2] Warrener, R. N.; Johnston, M. R.; Gunter, M. J. Synlett 1998, in press; Warrener, R. N.; Johnston, M. R.; Schultz, A. C.; Golic, M.; Houghton, M. A.; Gunter, M. J. Synlett 1998, in press.
[3] Warrener, R. N.; Elsey, G. M.; Maksimovic, Lj.; Johnston, M. R. Tetrahedron Lett. 1995, 36, 7753.
[4] Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P. J. Am. Chem. Soc. 1985, 107, 3902.
[5] Spartan v. 5.0., Wavefunction, Inc. 18401 Von Karman Suite 370, Irvine, California 92715, 1997.
[6] Roothan, C. C. J. Rev. Mod. Phys. 1951, 23, 69.
[7] Hehre, W. J.; Radom, L.; Schleyer, P.v.R.; Pople, J. ”Ab Initio Molecular Orbital Theory”, Wiley, N. York, 1986.
[8] Scharf, H.-D.; Droste, W.; Liebig, R. Angew. Chem. Int. Ed. Engl. 1968, 7, 215; Scharf, H.-D.; Kusters, W. Chem. Ber. 1972, 105, 564.
[9] Blakenspoor, R.; Chung, C. S. C., J. Org. Chem. 1975, 40, 2443.
[10] Warrener, R. N.; Russell, R. A.; Margetic, D. Synlett 1997, 38; Warrener, R. N.; Margetic, D.; Tiekink, E. R.; Russell, R. A. Synlett, 1997, 196; Warrener, R. N.; Elsey, G. M.; Houghton, M. A. Tetrahedron Lett. 1995, 36, 1417.
[11] Margetic, D.; Warrener, R. N. ECHET96, 24 June - 22 July 1996, presented as a poster.
[12] Wiest, O.; Montiel, D. C.; Houk, K. N. J. Phys. Chem. A 1997, 101, 8378; CPD+cyclic lactones: Sbai, A.; Branchadell, V.; Oliva, A. J. Org. Chem. 1996, 61, 621; CPD+maleic anhydride: Suarez, D.; Sordo, J. A. J. Chem. Soc. Chem. Commun., 1998, 385; CPD+maleimide, TAD (1,2,4-triazoline-3,6-dione): Xidos, J. D.; Poirier, R. A.; Pye, C. C.; Burnell, D. J. J. Org. Chem. 1998, 63, 105; substituted CPD + acrylonitriles: Domingo, L . R.; Picher, M. T.; Andres, J.; Vicent, S. J. Org. Chem. 1997, 62, 1775; substituted CPD dimerisations: Froese, R. D. J.; Organ, M.G.; Goddard, J. D.; Stack, T. D. P.; Trost, B. M. J. Am. Chem. Soc. 1995, 117, 10931; CPD+CPD: Jorgensen, W. L.; Lim, D.; Blake, J. F. J. Am. Chem. Soc. 1993, 115, 2936; Furan+cyclopropene: Jursic, B. S. Tetrahedron Lett. 1997, 38, 1305.
[13] Jursic, B. S. Computing Transition State Structures with Density Functional Theory Methods, in: Recent Developments and Applications of Modern Density Functional Theory; Seminario, J. M.; Ed.; Elsevier, Amsterdam, 1996, 709.
[14] Fukui, K.; Fujimoto, H. Bull. Chem. Soc. Jpn. 1967, 40, 2018; Fukui, K.; Fujimoto, H. Bull. Chem. Soc. Jpn. 1969, 42, 2018; Fukui, K. Angew. Chem. Int. Ed. Engl. 1982, 21, 801.
[15] Sauer, J.; Wiest, H. Angew. Chem. Int. Ed. Engl. 1962, 1, 269; Sauer, J. Angew. Chem. Int. Ed. Engl. 1967, 6, 16.
[16] Mulliken, R. S. J. Chem. Phys. 1955, 23, 1833; 1841; 2338; 2343.
[17] Carpenter, J. E.; Weinhold, F. J. Mol. Struct. (Theochem) 1988, 169, 41.
[18] Houk, K. N.; Li, Y.; Evanseck, J. D. Angew. Chem. Int. Ed. Engl. 1992, 31, 682.
[19] Margetic, D. Warrener, R. N. paper in preparation.
[20] Houk, K. N.; Loncharich, R. J.; Blake, J. F.; Jorgensen, W. L. J. Am. Chem. Soc. 1989, 111, 9172.
[21] Similar effect was found in TS with several other dienes: Beno, B. R.; Houk, K. N.; Singleton, D. A. J. Am. Chem. Soc. 1996, 118, 9984; Jorgensen, W. L.; Lim, D.; Blake, J. F. J. Am. Chem. Soc. 1993, 115, 2936; McCarrick, M. A.; Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc. 1993, 115, 3330; Sbai, A.; Branchadell, V.; Oliva, A. J. Org. Chem. 1996, 61, 621; Froese, R. D. J.; Organ. M. G.; Goddard, J. D.; Stack, T. D. P.; Trost, B. M. J. Am. Chem. Soc. 1995, 117, 10931.
[22] Cooley, J. H.; Williams, R. V. J. Chem. Ed. 1997, 74, 582.
[23] Loncharich, R. J.; Brown, F. K. Houk, K. N. J. Org. Chem. 1989, 54, 1129; Birney, D. M.; Houk. K. N. J. Am. Chem. Soc. 1990, 112, 4127.
[24] Jursic, B. S.; Zdravkovski, Z. J. Chem. Soc., Perkin Trans. 2 1995, 1223.
[25] Newmann, M. S.; Addor, R. W. J. Am. Chem. Soc. 1955, 77, 3789; Kwart, H.; Vosburgh, W. G. J. Am. Chem. Soc. 1954, 76, 5400.
[26] Anderson, W. K.; Dewey, R. H. J. Am. Chem. Soc. 1973, 95, 7161; Kowarski, C. R.; Sarel, S. J. Org. Chem. 1973, 38, 117.
[27] Haq, M. Z. J. Org. Chem. 1972, 37, 3015.
[28] Holthausen, M. C.; Koch, W. J. Phys. Chem. 1993, 97, 10021.
[29] Antol, I.; Eckert-Maksic, M.; Margetic, D.; Maksic, Z. B.; Kowski, K.; Rademacher, P. European Journal of Organic Chemistry 1998, in print.
[30] Camps, P.; Font-Bardia, M.; Mendez,
N.; Perez, F.; Pujol, X.; Solans, X.; Vazquez, S.;
Vilalta, M. Tetrahedron 1998, 54, 4679.