| [Related articles/posters: 089 023 119 ] |
Conjugated enamines are valuable intermediates in organic synthesis[1].
The marked increase of HOMO orbital energy respective to butadiene makes
enamines outstanding dienophiles in the "inverse electron demand" Diels-Alder
reaction[2].
It was reported[3], that dienamines can be
conveniently prepared by the ring opening of condensed heterocyclic salts.
We employed this strategy to the synthesis of tetrazolyl-dienamines and
-dienolethers (i.e. 2,3). Both 2 and 3
contain an electron rich diene chain and therefore reacted readily
with tetrazinedicarboxylic ester (4) to give tetrazolylvinylpyridazines
5 or 6 after the elimination of nitrogen and morpholine or
methanol[3]. The unexpected formation of the E-product
(5) from the Z-dienolether (3) prompted us to study the reaction
in detail, with special emphasis on the structure of the dihydropyridazine
intermediate (3tr) claimed to be responsible for the isomerisation[4].
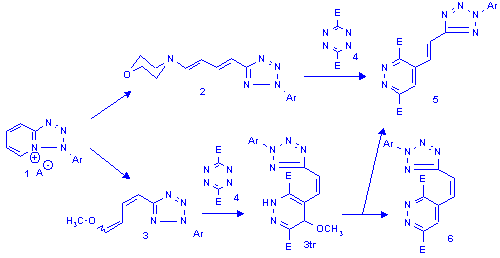
Results I.
To stabilise the 3tr structure a methyl group was introduced in b-position to the amine moiety through the 6-methyl-azolopyridinium salts (e.g. 7). The ring opening of 7 with pirrolidine afforded the E,E-dienamine 8 which reacted smoothly with two equivalents of tetrazinedicarboxylicacid diester (4) and two products were isolated which were identified as the tetrazolylpyridazine 9 and the dihydropyridazine 10. The unexpected[5] pathway shouldn't have gone through the analogue of 3tr as the attack of another molecule of tetrazine on 3tr is highly unlikely. Monitoring the reaction using 1H NMR measurements showed the presence of only unreacted starting material and two products at any stage of the process.
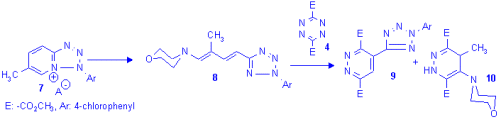
Hoping for a better insight, the reactions were also carried out using the less reactive 3,6-bis-(2'-piridyl)-tetrazine (11). There was no change in the reactivity pattern in the case of the methylated dienamine (8, R=CH3) and the pyridyl analogues of 9 and 10 were isolated (13, and 14 R=CH3) as the sole product, but the original dienamine (2) yielded minor quantities of the tetrazolylpyridazine 13 and the morpholino-dihydropiridazine 14 (R=H) besides the expected main product 12.
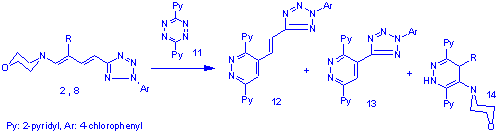
Rationalisation I.
The even charge and frontier orbital distribution[6] of dienamines suggests that the regiochemistry of the reactions is governed by steric factors. In the case of the nonmethylated dienamine the tetrazines attack preferentially at the enamine moiety and lead to dihydropiridazine intermediates (15) which readily aromatise to the azolylvinylpiridazines (12) by amine elimination. The introduction of a methyl group in b-position to the amine switches the site of preferable attack to the less substituted double bond next to the heteroaromatic moiety. The formed intermediate (16) still possesses an enamine moiety which makes it capable of reacting with a second equivalent of tetrazine to yield the bis-adduct (17). The disproportionation of 17 leads directly to the observed reaction products 13 and 14.
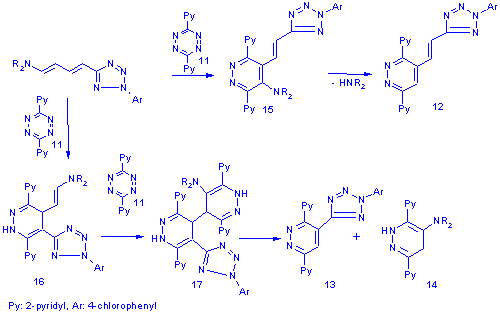
The 17 type intermediate is not unprecedented as Sandhu et al. proposed a similar double addition, carbon-carbon bond fission mechanism in their article[5] discussing the reaction of morpholino-butadiene with tetrazines.
Results II.
Testing the scope of the procedure a series of 1-amino-2-methyl-penta-1,3-dienes were reacted with tetrazinedicarboxylicacid diester (4) and in spite of the expectations the dienamines 18a,b (X=O, NMe) consumed only one equivalent of the tetrazine to give a single product in good yield which was identified as the new azo-bridged heterocycle 19a,b the structure of which has also been verified by X-ray crystallography[7] (19a).
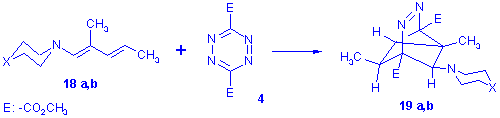
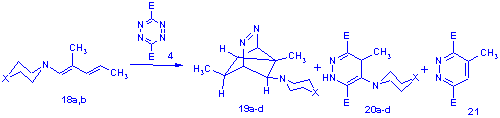
Efforts to extend the formation of 19 to other amine derivatives gave ambiguous results however, as examination of the crude products from the reaction of dienamines 18 a-d (X=O, NMe, CH2, nil) with 4 revealed a complex mixture of products. Besides a substantial amount of the tricyclo compounds 19a-d the dihydropyridazines 20a-d and the pyridazine compound 21 were also present in most cases, the amount of which seemed to be proportional to the basicity of the starting enamine[8].
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Rationalisation II.
We assume that steric factors govern the attack of the tetrazine molecule (4) to the D3,4-double bond of 18 and give the 2,5-dihydropyridazine 23 (the proposed tautomeric form is based on previous experience). The intermediate 23 can: (i) react with another molecule of tetrazine (4) to yield the bis-adduct 24 which on disproportionation gives 21 and 20 or (ii) tautomerise to the conjugated azdiene form and undergo an intramolecular Diels-Alder reaction leading to the tricycle 19.
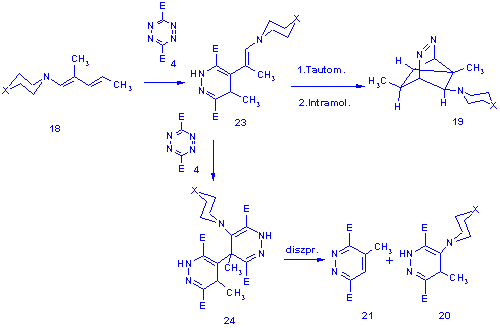
The more reactive enamines should in principle give higher yields of
the tricycle 19 through the conjugated azadiene tautomeric
form necessary for the second cycloaddition step. The opposite trend in
product distribution - e.g. the less reactive the enamine is, the higher
the proportion of the tricycle is - in the described cases suggests that
the crucial point in the process is the intermediateâs (23) ability
to tautomerise to the 4,5-dihydropyridazine form. With more reactive amines
the equilibrium is probably shifted strongly towards the 1,4-dihydropyridazine
form and the intermediate 23 is not consumed by the intramolecular
process and on meeting another molecule of tetrazine (4) undergoes
intermolecular Diels-Alder reaction, while in the cases of less reactive
amines the proportion of the conjugated 4,5-dihydropyridazine form is probably
high enough to promote the intramolecular addition and the formation of
19. Experimental support for the hypothesis comes from the finding
that slow addition of the tetrazine reagent to the dilute enamine solution
leads to a higher proportion of the tricycle in the product while ensuring
a large excess of the tetrazine by addition of the enamine to the tetrazine
solution results a considerable amount of bis adducts 20 and 21
(up to 50% with 18a).
Summary
Dienamines were shown to undergo selective transformations with tetrazine
derivatives and a series of products were isolated, The selective reactions
were rationalised by the steric non-equivalence of the two double bonds
in dienamines and the different tautomeric equilibria of the formed dihydropyridazine
intermediates.
References
[1] P.W. Hickmott, Tetrahedron, 1984,
40, 2989.
[2] For a reference on the reactivity of electron
rich olefins in the "inverse electron demand" Diels-Alder reaction see:
F. Thalhammer, U. Wallfahrer, J. Sauer, Tetrahedron Lett., 1990,
31, 6851 and references therein.
[3] A. Kotschy, Gy. Hajós, A. Messmer, J.
Org. Chem., 1995, 60, 4919.
[4] A similar isomerisation was also observed with
Z,E-azinyldienamines. See: A. Kotschy, Gy. Hajós, G. Timári,
A. Messmer, J. Org. Chem., 1996, 61, 4423.
[5] D.R. Borthakur, D. Prajapati, J.S. Sandhu, Heterocycles,
1987, 26, 337.
[6] Calculated net atomic charges and HOMO pp coefficients
(AM1, MOPAC 6.0) on the sp2-carbon atoms of 1-morpholino-1,3-pentadiene:
(a-d) q: -0.05, -0.22,
-0.12, -0.17; Cpz: -0.38, -0.49, 0.29, 0.44.
[7] A. Kotschy, D.M. Smith and A.Cs. Bényei,
Tetrahedron Lett., 1998, 39, 1045; a similar
reaction leading to the same novel ring system was published by T. Klindert,
P von Hagel, L. Baumann, G. Seitz, J. prakt. Chem., 1997,
339, 623.
[8] For a reference on the basicity of enamines see:
A.G. Cook, M.L. Absi, V.K. Bowden, J. Org. Chem., 1995,
60, 3169.