Drug Design and Discovery: 11,12
Salbutamol was developed from the modification of Norepinephrine, a natural neuro transmitter.
Norepinephrine: 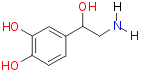
Norepinephrine stimulates a and b
adrenoceptors in the body, a more specific drug was need that targeted
only the b adrenoceptors.
It was found that the replacement of one hydrogens on the amine with
a isopropyl group produced isoproterenol (isoprenaline) which has increased
stimulation of b adrenoceptors whilst a reduced
stimulation of a adrenoceptors. However Isoproterenol
was not metabolically stable and was inactivated by the enzyme catechol
O-methyltransferase too quickly to be usefully used in the treatment
of asthma. Another disadvantage was that there are two types of b
adrenoceptors,
b1
and b2, isoproterenol activated both receptors
receptors. The activation of b1 adrenoceptors
in the cardiovascular system gave rise to side effects such as palpitations
and cardiac arrhythmia.
Isoproterenol: 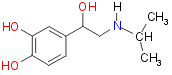
Thus salbutamol was developed (1969 Cullum et al10) by the replacement of an isopropyl group with a t-butyl group ( highlight the t-butyl group [molecule - top left hand corner]) and one alcohol group on the aromatic ring was replaced by a methanol group ( highlight the methanol group). Salbutamol also only stimulates b1 adrenoceptors but is also metabolically stable with an active time of about six hours in the body. Back to the original molecule.
Salbutamol: 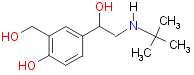
THE SYNTHESIS OF R SALBUTAMOL:14
Due to the dramatically different biological activity of the two different optical isomers of salbutamol the synthetic routes reported in the literature concentrate on the synthesis of R-salbutamol. These same syntheses can however with a suitable modification (e.g. change of resolution technique or catalyst) be used to produce the S isomer. Salbutamol is still commonly marketed as the less biologically effective racemic mixture which can be synthesised using the same routes without the diastereomeric resolution or asymmetric steps. There are many different approaches to obtaining pure R-salbutamol.
CONTENTS
1. Syntheses of R-salbutamol from a racemic mixture:
a) Via resolution of benzyl protected
arylethanolamine ester:
b) Via arylethanolamine methyl ester
(with Naproxen Resolution):
2. Asymmetric synthesis of R-salbutamol:
3. Synthesis of R-Salbutamol from R-Cyanohydrines:
A cheap and easy way to make R-salbutamol is to synthesis a racemic mixture and separate the isomers (either at an intermediate stage or of the final products). There is generally no exceedingly interesting chemistry involved in preparation of racemic mixtures to be resolved. The syntheses follow the general scheme of the addition of the requisite groups with protection of the more reactive groups to prevent their conversion.
1a) Via resolution of benzyl protected arylethanolamine ester:
Developed by Collin, DT et al15
Step 1:
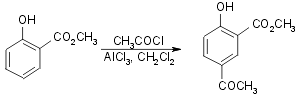
Yield : 70-75%
Step 2 + 3:

2 Steps: Total Yield 70%
Step 4:
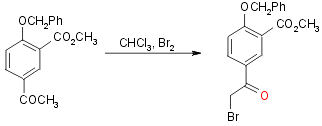
Yield 40%
Step 5: protected arylethanolamine ester:
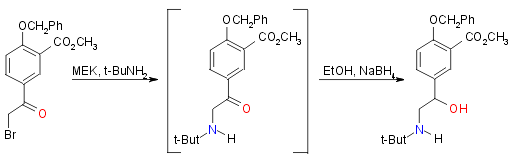
Yield 72%
(+)-DBTA Resolution: Harteley, et al16
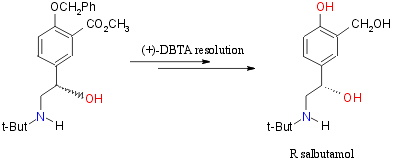
Total yield of (+)-DBTA Resolution 30-40%. Greater than 99.5% pure R-salbutamol
The process is advantageous as the process are short and efficient as
well as using non hazadous materials.
Debenzylation can be achieved under catalytic hydrogen transfer and
the process uses commercially available intermediates.
1b) Via arylethanolamine methyl ester (with Naproxen Resolution):
Yield 70-75%
Step 2:
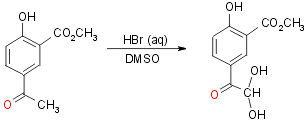
Yield 80% Crude (aprox)
Step 3:
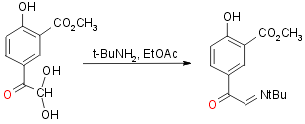
Yield 75-82%
Step 4:
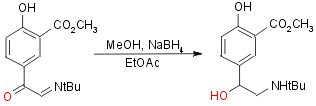
Yield 70%
Step 5 + 6:
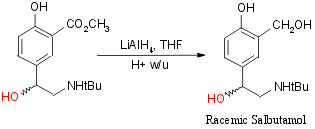
Step 7+8:
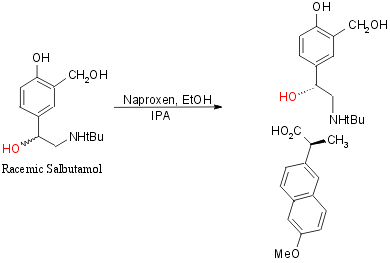
Naproxen: (+)-6-Methoxy-a-methyl-2-naphthaleneacetic
acid, CH3OC10H6CH(CH3)CO2H
Two steps, Total Yield 15%, 86.4%cc
Step 9:
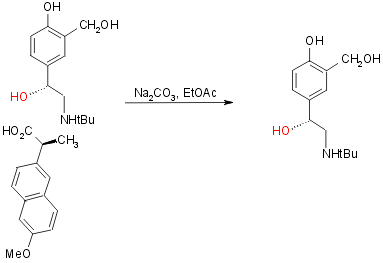
Many different resolution targets can be used to produce pure R-salbutamol including, as in the second synthesis, a racemic mixture of salbutamol its self. For instance:
- DTTA resolution16 of dibenzyl protected arylethanolamine ester
Diasteriomeric resolutions, whilst having good yields, can be long and tedious. Thus more recently routes to R-salbutamol have been developed using asymmetric reduction methods (of a-ketoimines or a-bromoketones).
Asymmetric reduction of a-ketoimine:
First Reported in 1994 by Hong et al17
the synthesis of the relevant a-ketoimine can
be found above (steps 1-3 of the second synthesis: go
there)
The reduction route was in 1988 by kitamura et al18
The reaction is:
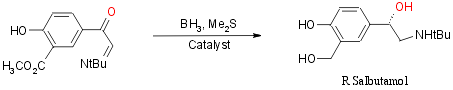
This reaction is catalyzed by oxazaborolidines. The reduction with the
boro hydride results in a new chiral center forming and with the right
choice of catalyst the desired stereo isomer can be chosen. The chiral
catalysts are stereo specific.
Catalysts for this reaction include:
I 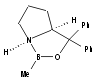 Developed
by Corey19 and the Merck
group21
Developed
by Corey19 and the Merck
group21
II a 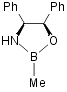 Pfizer catalyst22
Pfizer catalyst22
II b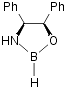 Pfizer
catalyst22
Pfizer
catalyst22
III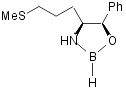 Martens
catalyst23
Martens
catalyst23
IV 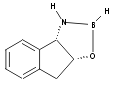
(Models were generated using Chem3D pro and MM2 minimisation.)
The yield of this reaction is greatly effected by the catalyst used,
the temperature, the solvent used and the length of time over which addition
of the reducing agent occurs. The best enantiomeric excess can be achieved
using catalyst I at 100% M concentration, in toluene, 0oC, and
a slow (over 3 hour) addition time. This gave an ee of 93-95% with a greater
than 99% conversion.
This same method can also be used to reduce the benzyl protected a-ketoimine.
The optimum conditions for this reduction are: a slow addition (<3hr)
in toluene at -10oC. This gives a 91% yield (99% ee 99.7% purity)
This method gives excellent yield and very pure products without the tedious diasteriomeric resolution. However using 100% M concentrations of catalyst is not very practical industrially, (although excellent yields (90%+ are achieved with lower concentrations).
Another synthesis of R-salbutamol reported recently in the literature
is that starting the corresponding R-cyanohydrin. This was reported by
Effenberg et al24
This introduces a N-tert-butyl group via the Ritter reaction followed
by hydrogenation and deprotection The overall reaction is:
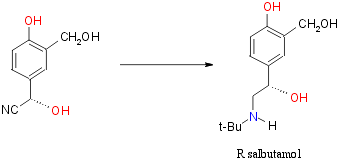
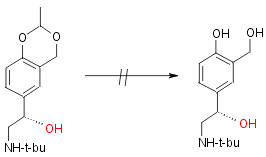
However at the present time the deprotection step (hydrolytic cleavage
of the acetal) has not been successfully achieved without racemization
and partial decomposition.
The reaction scheme is:
Step 1:
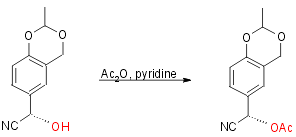
Yield 92%
Step 2:
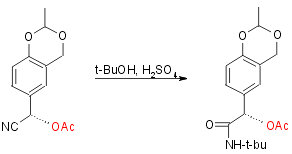
Yield 62%
Step 3:
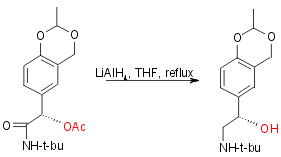
Yield 85%
Step 4: Not yet synthetically possible
Although Salbutamol was first synthesised in 1969 there is still much research into better, quicker and more efficient syntheses of R-Salbutamol. There is currently only one major synthesis of R-Salbutamol although there are many possible ways of producing pure samples of R-Salbutamol via diastereomeric resolution.