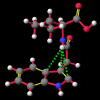
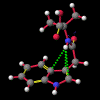
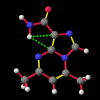
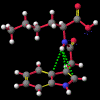
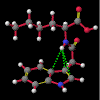
"Non-classical" hydrogen bonding interactions may represent important terms in molecular and chiral recognition [1]. Previous work [2] carried out by this group to identify compounds that exhibit this behaviour has evolved into a program of modelling the range of structural characteristics tolerated, ultimately with the intention of designing systems to take advantage of such effects. In this paper, a seminal early report by Burley and Petsko [3] that a number of enzymes exhibited unusually close NH approaches to the face of phenyl rings is followed up with an analysis of a small molecule search of the Cambridge Crystallographic Database [4] for NH...pi-facial distances significantly shorter than the van der Waal's contact distances. The search revealed a total of fifty-two structures. In each of the original reports, these features were either attributed to crystal packing effects or else overlooked entirely. Of particular interest, amongst an otherwise diverse selection, were a group of indole-3-acetic acid (auxin) analogues that had been synthesised to investigate the effects of conformation on plant-growth promoting activity.
Earlier studies had suggested that there was some correlation between the flexibility and orientation of the side chain with respect to the indole ring and the biological activity. These factors were, at the time, felt to be predominantly influenced by intermolecular hydrogen bonding. However, conformational energy calculations carried out using AM1 and PM3 semi-empirical methods, supported by 6-31G(d, p) ab initio and 6-31(d) density functional computations, would suggest that there is also a significant (~1-2 kCal/mol) intramolecular NH...pi-facial hydrogen bonding interaction, which is of primary importance in determining the alignment of the substituent.
Theoretical calculations were carried out on these at the restricted Hartree-Fock (RHF) level using the AM1 and PM3 semi-empirical SCF-MO methods, as implemented in the MOPAC 93 [5] program. Structures were optimised using the Broyden, Fletcher, Goldfarb and Shanno method to a final gradient norm of less than 0.01. The parameters defining the SCF criterion and trust radius were set to 100M and -100 respectively and the MMOK keyword was invoked in the instance of a peptide bond. Conformational searches were then performed by selecting the sigma-bond that was closest to being parallel to the hypothetical NH...pi-facial interaction and calculating the heats of formation as the molecule was twisted through 360° in 5° steps about this axis. Invariably, distinct minima could be found. By taking the difference in energies between these minima, it was possible to obtain some measure of the strength of the hydrogen bonding interaction.
For the systems of particular interest that will be described later in this paper, model structures were designed which were subjected to ab initio optimisation and analysis using the Gaussian 92/DFT revision G2 [6] suite of programs. Optimisations were carried out using the 6-31G** basis set with Hartree-Fock and the 6-31G* with Becke, Lee, Yang and Parr Density Functional methods.
Molecular diagrams were generated and inspected in true 3D stereo using both CAChe Scientific and Silicon Graphics Indigo workstations, the latter running locally developed EyeChem modules [7] within the IRIS Explorer 3.0 environment.
As has been discussed in the previous paper [2], the distance defining a significant hydrogen bonding interaction is somewhat arbitrary. With this work, it was again decided that intra-molecular interactions significantly (0.2 - 0.6 Å) shorter than the van de Waal's contact distances be focussed on.
The initial database search, specifying a contact distance of between 1.5 and 2.5 Å from an amine hydrogen to both carbon atoms in a C=C system gave five hits. In two of these, the close contact had obviously been 'forced' by a highly strained ring system, and in another two the cause of the interaction could not easily be attributed due to their excessive size. The remaining structure - WEBBAG - was retained for further computational analyisis. The contact distance was then redefined as being between 1.4 and 2.4 Å from an amine hydrogen to either carbon atom in a C=C system. This search proposed fifty-two structures, including the one retained from the first search. Of these, thirty-one were discarded on the grounds that they were either too large for detailed computational analysis or else contained metallic centres, although it was noted that, in twenty-one of these, the short interaction was to a porphyrin system. In several other cases, the short NH...pi distance may be attributed to factors other than a hydrogen bonding interaction, being the result either of a severely distorted ring system or a nearby carboxy anion, and these too were rejected. The remaining fourteen structures were all deemed to be suitable: the positions of the hydrogen atoms had been refined and the R-factors were acceptably small, at just under 8% in the worst case and with most others falling below 5%.
Table 1 shows the Cambridge NH...C bond distances (the two numbers quoted being the distance from the hydrogen to each of the C=C carbons) for each structure together with the corresponding calculated distances and heats of formation of the semi-empirically optimised structures. The estimated hydrogen bond strengths are also given.
| Refcode: | r NH...C (Expt)* | R-factor | Method | Heat of Formation (kCal/mol) | r NH...C (Calc)* | Calc H-Bond Energy(kCal/mol) |
|---|---|---|---|---|---|---|
| BEGRIO | 2.346 3.244 | 0.046 | AM1 | 86.353 | 2.531 3.587 | 2.39 |
| PM3 | 53.932 | 4.316 5.366 | 2.78 | |||
| FRUTIC | 2.386 2.850 | 0.062 | AM1 | -15.584 | 2.597 2.656 | 2.37 |
| PM3 | -34.808 | 2.724 3.385 | 1.63 | |||
| JIMZAG | 2.437 2.514 | 0.041 | AM1 | -99.607 | 2.588 2.787 | 0.53 |
| PM3 | -115.721 | 2.583 2.797 | 0.00 | |||
| KACKII | 2.381 3.428 | 0.065 | AM1 | 38.541 | 2.591 3.660 | 1.75 |
| PM3 | 11.146 | 2.953 4.164 | 0.76 | |||
| KIHYUV | 2.393 3.071 | 0.048 | AM1 | -83.752 | 2.603 3.642 | 0.40 |
| PM3 | -98.577 | 2.730 3.786 | 0.82 | |||
| KIHZAC | 2.368 2.995 | 0.045 | AM1 | -90.060 | 2.603 2.643 | 0.49 |
| PM3 | -103.079 | 2.727 3.782 | 1.10 | |||
| KIHZEG | 2.285 2.991 | 0.065 | AM1 | -96.865 | 2.602 3.641 | 0.51 |
| PM3 | -108.479 | 2.728 3.784 | 1.09 | |||
| KIHZIK | 2.206 3.017 | 0.078 | AM1 | -174.143 | 2.590 2.549 | 1.27 |
| PM3 | -185.013 | 2.742 2.707 | 0.60 | |||
| LACCAT | 2.355 2.945 | 0.052 | AM1 | -94.405 | 2.598 3.640 | 0.46 |
| PM3 | -107.237 | 2.702 3.758 | 1.20 | |||
| LACCEX | 2.357 2.960 | 0.042 | AM1 | -83.323 | 2.534 2.554 | 0.03 |
| PM3 | -101.169 | 2.601 2.633 | 1.08 | |||
| PECVAU | 2.263 2.282 | 0.021 | AM1 | 45.796 | 2.612 2.777 | 0.35 |
| PM3 | 7.739 | 2.591 2.887 | 0.49 | |||
| VOXHUL | 2.394 3.145 | 0.072 | AM1 | -103.695 | 2.600 3.638 | 0.50 |
| PM3 | -113.885 | 2.717 3.770 | 1.14 | |||
| WEBBAG | 2.477 2.492 | 0.038 | AM1 | 157.55 | 2.525 2.542 | 1.25 |
| PM3 | 132.348 | 2.562 3.236 | 0.51 |
Of the fourteen structures, it is remarkable that in ten the close contact was to the C3 of an indole system. In the case of WEBBAG, the hydrogen is located symmetrically above the C2=C3 bond. With the other nine it is closest to the C3, straddling the -C2=C3-C31= section and pointing towards the indole nucleus. WEBBAG is perhaps the best example of the semi-empirical methods' shortcomings in predicting the existence of such hydrogen bonds, with the PM3 calculation hopelessly overestimating the contact distance. The AM1 calculation, by contrast, comes within 0.05 Å of the experimental measurement. The estimate of 1.25 kCal/mol can therefore be regarded as being a lower limit for the interaction energy in this case.
This behaviour - namely that AM1 outperforms PM3 in predicting the structures of these molecules, but still overestimates the contact distance of the NH to the C=C double-bond - can be observed to a greater or lesser degree in all of the systems in question. It should be noted that in none of the examples considered does the NH bear directly down onto the C=C bond, and it would be expected that, in systems expressly designed to exploit this effect, shorter, higher energy interactions will be observed. It can, however, be concluded from these semi-empirical studies on actual experimental data that while the calculated interaction energies must be regarded with a good deal of caution quantitatively, they nevertheless suggest a small but significant effect.
Indole-3-acetic acid (IAA; auxin) was identified as long ago as 1934 as having plant growth promoting properties. Although it has been established that it regulates such physiological functions as cell division and enlargement, protein synthesis and developmental differentiation [8], its mode of action is still the subject of much debate. It has, however, been noted [9], [10] that bound auxins or auxin conjugates are involved in hormone transport, and serve as long- and short-term storage forms of hormone.
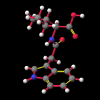
In order to shed some light on the subject of molecular recognition of auxin and its biologically active conjugates, a Croatian group embarked on a programme of synthesising N-indol-3-ylacetyl derivatives of aliphatic amino acids [11], determining their crystal structures and attempting to correlate their structural and physico-chemical parameters - the relative orientation of side chains towards the indole nucleus; the intramolecular distance between the active sites (known to be the indole nitrogen and the C=O); the distribtuion of the hydrophobic and hydrophilic regions and the electronic propereties of the aromatic system - with their biological activity.
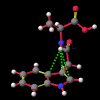
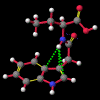
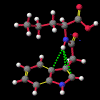
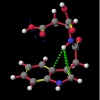
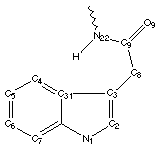
A number of these structures were picked up in the search of the Cambridge Crystallographic Database. The additional structures KIHZUW, KIJBAG, KUBMEZ, KUBMID and KUBMOJ were not found in the original database search, either because the crystal packing was affected by very long side chains, causing the C9=O9 to point towards the indole centre, or else because the NH...C contact was very slightly greater than 2.4 Å. They were, however, discussed in the original papers and were found by a search for the indole-3-acetamide fragment. The atom numbering scheme employed in this work (see figure) is that used throughout in the reports by Kojic-Projic, et al., and has been retained so as to simplify cross-referencing.
|
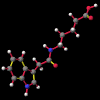
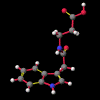
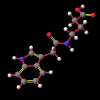
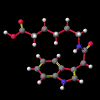
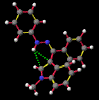
A significant finding in these studies was that there was some relationship between the C9=O9 group relative to the indole system. Certain anomalous results (KIJBAG, KUBMID) described in these papers - whereby the C9=O9 was pointing towards rather than away from the indole nucleus - were shown by NOE measurements in solution, and confirmed by molecular mechanics, to be entirely due to intermolecular hydrogen bonding and packing effects in the crystalline state, and therefore irrelevant to the functioning of the free molecule.
What was not seen, or at any rate discussed, was the remarkably short N22-H...C3 distance (< 2.4 Å), the fact that this hydrogen was pointing directly towards the centre of the indole system and that this distance shortened considerably as the molecule was cooled (VOXHUL vs. VOXHUL01). It could be argued that since, in any given HCNO peptide linkage, the oxygen is ideally antiperiplanar to the hydrogen, and so the relative orientation of the C9=O9 is dependent on that of the N22-H.
It is the contention here that the N22-H...C3 contact is due to an intramolecular hydrogen bond with the aromatic pi-system of the indole moeity. The semi-empirical optimised conformational energy searches show that this interaction varies in the region of 0.4-2.0 kCal/mol.
Table 2 shows the Cambridge NH...C bond distances (the three numbers quoted being the distance from the hydrogen to the C3, C2 and C31 carbons respectively) for each structure together with the corresponding calculated distances, heats of formation of the semi-empirically optimised optimised structures and the estimated hydrogen bond strengths as before.
| Refcode: | r NH...C (Expt)* | R-factor | Method | Heat of Formation (kCal/mol) | r NH...C (Calc)* | Calc H-Bond Energy(kCal/mol) |
|---|---|---|---|---|---|---|
| KIHYUV | 2.393 3.071 2.770 | 0.048 | AM1 | -83.752 | 2.603 3.642 2.590 | 0.40 |
| PM3 | -98.577 | 2.730 3.786 2.744 | 0.82 | |||
| KIHZAC | 2.368 2.995 2.733 | 0.045 | AM1 | -90.060 | 2.603 2.643 2.591 | 0.49 |
| PM3 | -103.079 | 2.727 3.782 2.769 | 1.10 | |||
| KIHZEG | 2.285 2.991 2.642 | 0.065 | AM1 | -96.865 | 2.602 3.641 2.590 | 0.51 |
| PM3 | -108.479 | 2.728 3.784 2.770 | 1.09 | |||
| KIHZIK | 2.206 3.017 2.507 | 0.078 | AM1 | -174.143 | 2.590 2.549 3.665 | 1.27 |
| PM3 | -185.013 | 2.742 2.707 3.901 | 0.60 | |||
| KIHZUW | 2.403 3.016 2.979 | 0.077 | AM1 | -101.234 | 2.622 3.690 2.585 | 1.69 |
| PM3 | -112.728 | 2.698 3.750 2.731 | 0.18 | |||
| KIJBAG | 3.750 4.970 4.288 | 0.054 | AM1 | -101.098 | 2.591 3.630 2.575 | 0.19 |
| PM3 | -110.245 | 2.741 3.814 2.777 | 1.82 | |||
| KUBMEZ | 2.430 3.185 2.814 | 0.036 | AM1 | -87.447 | 2.618 3.693 2.577 | 1.37 |
| PM3 | -99.532 | 2.758 3.850 2.787 | 1.93 | |||
| KUBMID | 3.817 5.010 4.394 | 0.030 | AM1 | -94.526 | 2.582 3.617 2.560 | 1.22 |
| PM3 | -104.000 | 2.614 3.610 2.699 | 2.90 | |||
| KUBMOJ | 2.405 3.055 2.830 | 0.048 | AM1 | -107.573 | 2.453 2.812 3.141 | 0.29 |
| PM3 | -115.779 | 2.450 3.029 2.921 | 1.31 | |||
| LACCAT | 2.355 2.945 2.807 | 0.052 | AM1 | -94.405 | 2.598 3.640 2.581 | 0.46 |
| PM3 | -107.237 | 2.702 3.758 2.726 | 1.20 | |||
| LACCEX | 2.357 2.960 2.848 | 0.042 | AM1 | -83.323 | 2.534 2.554 3.559 | 0.03 |
| PM3 | -101.169 | 2.601 2.633 3.695 | 1.08 | |||
| VOXHUL | 2.394 3.145 2.968 | 0.072 | AM1 | -103.695 | 2.600 3.638 2.588 | 0.50 |
| PM3 | -113.885 | 2.717 3.770 2.754 | 1.14 | |||
| VOXHUL01 | 2.320 3.068 2.642 | 0.053 | Voxhul at 133K |
There is an unfortunate discrepency between the optimal semi-empirical and experimental structures. Excepting KIHZIK and LACCEX, but including the additional five structures given by the search for the indole-3-acetamide moeity, it was observed that with the AM1 parameter set the hydrogen is generally located above the C3-C31 bond, whereas with the PM3 it is, if anything, pointing towards the C4. In the crystal structure, it bifurcates the C3-C31 and C3-C2 bonds, pointing roughly towards the centre of the indole nucleus. A Hartree-Fock 6-31G(d, p) ab initio calculation on a model system (indole-3-acetamide) had the hydrogen above the C3-C2, agreeing roughly with the semi-empirical calculations on KIHZIK and LACCEX. However, using the Becke, Lee, Yang and Parr density functional with a 6-31G(d) basis, an almost perfect agreement with the crystal structureswas obtained, suggesting that electron correlation effects are significant.
An identical DFT calculation on indole acetic acid itself again showed the short H-C3 distance, demonstrating once more that this conformation is preferred by the free molecule - intermolecular hydrogen bonding bringing about the contadictory crystal structure - this time being the result of an OH-pi interaction.
It is beyond the scope of this work to attempt to correlate the strength of the proposed hydrogen-bonding interaction with the biological activity, not least because of the trepidation involved when considering the semi-empirical energies. It is, however, being stated that the orientation of the carboxyl group in the free molecule is a direct consequence of this interaction.
Semi-empirical calculations are quite poor at predicting these interactions, with the AM1 parameter set performing marginally better across the range of molecules selected. This, too, is consistent with the findings of earlier work. Unfortunately, accurate modelling of these "non-classical" hydrogen bonds can, at present, only be achieved by performing ab initio calculations that consider both polarisation and electron correlation effects. The systems described might, therefore, also serve a valuable role in the evaluation of novel modelling methodologies.
1. P. Camilleri, D. S. Eggleston, H. S. Rzepa and M. L. Webb, J. Chem. Soc., Chem. Commun., 1994, 1135.
2. H. S. Rzepa, M. H. Smith and M. L. Webb, J. Chem. Soc., Perkin Trans. 2, 1994, 703.
3. S. K. Burley and G. A. Petsko, FEBS Lett., 1986, 203, 139.
4. F. H. Allen, J. E. Davies, J. J. Galloy, O. Johnson, O. Kennard,
C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson,
J. Chem. Inf. Comput. Sci., 1991, 31, 187.
See also: Cambridge Crystallographic Data Centre
5. M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. W. Wong, J. B. Foresman, M. A. Robb, M. Head-Gordon, E. S. Replogle, R. Gomperts, J. L. Andres, K. Raghavachari, J. S. Binkley, C. Gonzalez, R. L. Martin, D. J. Fox, D. J. Defrees, J. Baker, J. J. P. Stewart and J. A. Pople, Gaussian, Inc., Pittsburgh, PA, 1993.
6. J. J. P. Stewart,
Fujitsu Ltd., Tokyo, Japan, 1993.
Available from Quantum Chemistry Program Exchange, University of Indiana,
Bloomington, IN.
7. O. Casher, H. S. Rzepa and S. Green,
J. Mol. Graphics., 1994, 12, 226.
See also: EyeChem Modules
8. P. J. Davies, Plant Hormones and their role in Plant Growth and Development, Martinus Nijhoff, Dordrecht, 1987.
9. J. D. Cohen and R. S. Bandurski, Annu. Rev. Plant Physiol., 1982, 33, 403-430.
10. V. Magnus, in Conjugated Plant Hormones: Structure, Metabolism and Function (eds. K. Schreiber, H. R. Schuette and G. Sembdner), VEB Deutscher Verlag der Wissenschaften, Berlin, 1987.
11. B.Kojic-Prodic, B.Nigovic, D.Horvatic, Z.Ruzic-Toros, V.Magnus, W.L.Duax,
J.J.Stezowski, N.Bresciani-Pahor,
Acta Cryst.,B (Str.Sci.), 1991, 47, 107.
B.Nigovic, B.Kojic-Prodic, V.Puntarec, J.D.Schagen,
Acta Cryst.,B (Str.Sci.), 1992, 48, 297.
B. Nigovic, B. Kojic-Prodic, V. Puntarec,
Acta Cryst.,C (Cr.Str.Comm.), 1992, 48, 1079.
B.Kojic-Prodic, B.Nigovic, V.Puntarec, S.Tomic, V.Magnus,
Acta Cryst.,B (Str.Sci.), 1993, 49, 367.