Published in J. Chem. Soc., Perkin Trans. 2, 1994, 703.
(c) Royal Society of Chemistry, 1994.
A Crystallographic and PM3 SCF-MO Investigation of Strong OH... [pi]-Alkene and Alkyne Hydrogen Bonding Interactions
Henry S. Rzepa,[a] Mark H. Smith and Michael L. Webb[b]
Department of Chemistry, Imperial College of Science
Technology and Medicine, London, SW7 2AY, U.K. E-mail: rzepa@ic.ac.uk,
[b] SmithKline Beecham Pharmaceuticals, Old Powder Mills, Leigh,
Tonbridge, Kent, TN11 9AN.
Summary. A search of the
Cambridge structural database reveals eleven alkenes and two alkynes in which
particularly short OH...[pi]-facial hydrogen bonding occurs, a feature largely
unrecognised in the original reports of these structures. A wide structural
diversity is found for these structures, and in two cases, the [pi]-facial
bonding may be associated with anomalous reported reactivity. Semi-empirical AM1
and PM3 SCF-MO calculations tend to overestimate the C...H distances in these
structures, and very probably under-estimate the energies of OH...[pi]
interaction.
Although spectroscopic evidence, mostly based on
observed shifts in infra-red spectra, for [pi]-facial interactions with
hydroxyl groups has long been available,[1] crystallographic
evidence has been restricted to a small number of isolated reports of individual
compounds[2]. A recent survey[3] based on
the 1990 version of the Cambridge structural database[4]
containing 82129 structures suggests that the phenomenon
may be more widespread than hitherto recognised. However, this reported survey
of the literature did not extend to discussion of individual compounds or to
whether any common structural features can be discerned. Since such weak
interactions may represent important terms in molecular and chiral
recognition,[5] it is important to identify the range of
structural characteristics tolerated, and to assess whether standard theoretical
procedures correctly reproduce these interactions.
At the molecular
mechanics level of modelling, specific geometrical functions are often used in
mechanics force fields to describe hydrogen bonding, and the unusual
characteristics of [pi]-facial interactions may fall outside the range of
these functions. Recent advances [6] in using a distributed
multipole analysis of the molecular charge distribution hold considerable
promise for reproducing such non-classical hydrogen bonds, and it is therefore
highly desirable to identify a number of specific examples for use in the
calibration of various modelling methods. Semi-empirical SCF-MO based methods
have not hitherto been evaluated for these types of interactions. Amongst these
methods, the MNDO parametrisation performs poorly regarding both geometrical
energetic predictions for hydrogen bonds involving oxygen or nitrogen.[6]
The AM1 method represents a significant improvement in
terms of the energetics of such interactions, although the predicted geometries
tend towards biscected structures, whilst the most recent PM3 method appears to
most consistently reproduce the dimensions of bonding to oxygen.[7]
To provide a set of geometrical data against which to
evaluate the characteristics of these methods, we performed a search of the
109992 crystal structures in the current (March 1993) version of the Cambridge
database in which we identify thirteen compounds which exhibit particularly
short OH...[pi]-facial interactions. In the present paper we discuss these
structures individually in order to establish if any common structural themes
can be identified, and to clearly identify a set of compounds against which
modelling techniques can be tested.
Computational Details. Theoretical calculations were carried out at the restricted Hartree-Fock
level (RHF) using the AM1 or PM3 semi-empirical SCF-MO methods, as implemented
in the MOPAC 93 program.[8 ] with the MMOK keyword enabled.
Structures were optimised using the eigenvector following algorithm to a final
gradient norm of < 0.1, a tolerance required to obtain reproducible distances
for weak interactions. The search of the Cambridge structural database was
performed using release 5.5 of the Quest software. Structures are identified in
the text below using the Cambridge REFCODES. Structures for which the original
authors have noted that the hydroxyl hydrogens positions were refined are
indicated in the Table. All the R factors were acceptably small. Molecular
diagrams were generated and inspected in true 3D stereo using a CAChe Scientific
workstation. Computer readable files for Apple Macintosh and Microsoft Windows
systems in Quicktime(TM) video animation format illustrating in colour the three
dimensional properties of the structures in the Table are available for general
access from the Gopher+ server gopher.ch.ic.ac.uk. These files will
reside in the Royal_Society_of_Chemistry/Perkin_Transactions_2/3_05613L
directory for a period of at least two years from the publication of this paper.
A description of how to visualise such material, together with appropriate
programs is available from the same source.
Results and Discussion. The distance defining significant hydrogen bonding
interactions is to some extent arbitrary. Several recent reports[2] of [pi]-OH bonding to an aromatic ring suggest the
shortest contacts range between 2.2 to 2.5Å for the approach of a hydrogen
atom and the plane of the ring, compared with an estimate of the combined H and
C van der Waals radii of ~2.8Å. Desiraju and co-workers [3]
chose a criterion which resulted in a mean intermolecular
H...C distance of 2.69Å to alkynes and 2.77Å to alkenes, values
which are quite similar to the combined atomic van der Waals radii. There must
remain some ambiguity about whether H...C distances of > 2.8Å are
predominantly dispersive or electrostatic in character. The very shortest of
such interactions were not identified by Desiraju and co-workers. [3]
In our own search, we had independently chosen to focus on
both intra and intermolecular interactions significantly shorter (0.2 -
0.6Å) than the van der Waals contact distances. We first defined our
search of the Cambridge database as any intra or intermolecular contact of a
specified distance between an O-H proton and both carbon atoms of a C=C
or C[equivalence]C bond. A value of <= 2.5Å produced five hits, with
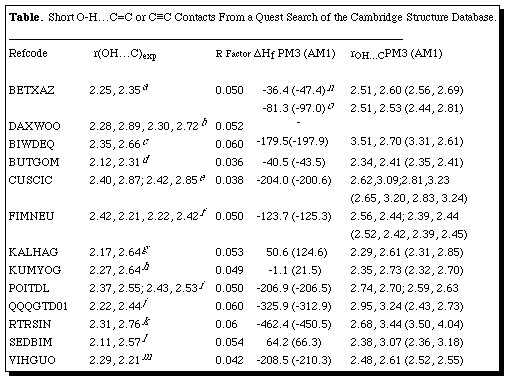
Cambridge "REFCODES" of BETXAZ, BUTGOM, FIMNEU, QQQGTD01 and VIHGUO. Specifying
that only one carbon atom need be within 2.4Å of the hydrogen gave more
hits (40), but a significant number of contacts are to enolic type alkenes and
are not discussed here, as are two (BREFEL and FEJHOR) where the proton position
was clearly mis-assigned in the original refinement.
Thirteen structures
appear to exhibit OH...[pi]-facial interactions significantly shorter than the
van der Waals contact distances (Table, Figure), of which four are
intermolecular and the others intramolecular. The previous search of the 1990
database[4] using version 4 of the Quest software was
restricted to searching only for intermolecular contacts. Only one of the
OH...[pi] interactions was correctly identified in the original papers
reporting the structures (that for QQQGTD01). Another
(BETXAZ) was mis-assigned to an intramolecular interaction (vide infra).
This suggests that the phenomenon has not been clearly identified in the past,
in part because the molecular structure and stereochemistry is frequently the
principal reason for x-ray characterisation, rather than the detail of the
intermolecular contacts.
Two of the short intermolecular
examples both involve alkynes. The structure of BETXAZ reveals a core
dimeric [pi] C[equivalence]C...HO interaction (erroneously assigned to be
intramolecular in the original report, Table 1), augmented by additional
terminal C[equivalence]C-H...O interactions from two further molecular
units.[9] This highly unusual structure is probably promoted
by the hydrophobic and sterically constraining adamantyl group inhibiting more
conventional O..HO interactions. We note in passing that similar simultaneous
terminal C[equivalence]C...metal and [pi] C[equivalence]C...Na+ ion interactions have also been reported[10].
Hanton and Hunter have also recently discussed other
possible reasons for the promotion of non-classical hydrogen bonding
interactions.[11 ] The phosphine-platinum complex DAXWOO
reveals an unusual bifurcated interaction between water and an [alpha]-hydroxy
alkyne in which the hydrogen bond appears to form to both the oxygen and one
carbon of the alkyne. Given the presence of a heavy metal atom close to the
refined proton position, this should be a good candidate for a more accurate
structure determination to verify this interaction.
Only two short
intermolecular examples to alkenes were found (RTRSIN and BIWDEQ), the
latter to a trans double bond present in a 9-membered ring. No
monosubstuted alkene examples were found, in accord that such alkenes are
normally regarded as being less basic than more heavily substituted systems.
Surprisingly, four alkenes bearing conjugated substituents were identified,
CUSCIC and VIHGUO to a carbonyl group, SEDBIM to one coplanar phenyl group and
FIMNEU as a diene. It appears that conjugation does not reduce the basicity of
the [pi]-bond sufficiently to inhibit such bonding. In the first three
examples, the hydrogen bonding is localised largely to the alkene, but
remarkably in FIMNEU the OH group has an equal attraction to both double bonds.
In CUSCIC and POITDL, an additional intermolecular OH...O hydrogen bond is
present at one or both [pi]-bonding centres (Figure). A number of the
structures show an essentially symmetric interaction with the [pi]-system
(< 0.25Å difference between the two C...H distances, cf BETXAZ,
BUTGOM, FIMNEU, POITDL, QQQGTD01 and VIHGUO) whilst in the remaining structures,
the asymmetry is closer to 0.4Å (Table). In all the intramolecular
examples except SEDBIM, the proximity of the OH and alkene is enforced
structurally, although the O-H bond is potentially free to rotate away from the
double bond and hence to form intermolecular OH...O interactions.
For MPEG animations of the various structures, click on any entry below.
BETXAZ , DAXWOO ,BIWDEQ
,BUTGOM ,CUSCIC ,FIMNEU ,
KUMYOG ,KALHAG ,POITDL ,
QQQGTD01 RTRSIN,
SEDBIM , VIHGUO
In two instances, it is noted in the original structural report that unexpected
reactivity was observed. For example, BUTGOM was observed to cyclise on standing
to an ether, and for SEDBIM intramolecular photochemical H-transfer occurs in
the solid state (Table). In neither case was an explanation suggested, but the
association of unexpectedly high reactivity and [pi]-facial hydrogen bonding
may provide one such explanation. Clearly, other instances of anomalous
reactivity may come to light if the phenomenon is more widely recognised.
Theoretical calculations. The semi-empirical calculations
reveal that in general these methods tend to overestimate the length of the
[pi]-C...HO interactions, with broadly similar behaviour between the AM1 and
PM3 methods. For the intermolecular interaction in BETXAZ, we calculated both
the dimeric unit, and the extended tetramer which also includes an additional
terminal C[equivalence]C-H...O interaction. In the dimer, the H...C distance
is ~0.3Å too long at both the AM1 and PM3 levels, although the asymmetry
in the [pi]-interaction is reproduced well (Table). In the calculated
tetramer, the additional polarisation induced by the terminal C-H...O
interaction [rCH...O 2.26Å, rCH...O 1.83Å (PM3), rCH...O 2.26Å
(AM1)] has only a slight effect on the calculated [pi] C[equivalence]C...HO
distances (Table), with the asymmetry being increased with AM1, and decreased
with PM3. The other intermolecular examples (BIWDEQ and RTRSIN) do not show
significantly different behaviour from the remaining intramolecular examples.
Several of the intramolecular systems (FIMNEU, KALHAG, KUMYOG) reveal a quite
close correspondance between calculated and observed structures. The energy of
the [pi]-interaction orientation in FIMNEU is calculated to be 1.3 kcal mol-1 lower at both the AM1 and PM3 levels than the rotamer
involving no [pi]-facial interaction, a value which appears on the low side,
and comensurate with the over-estimated bond lengths. The corresponding
dimerisation energies for the triple bonded system BETXAZ are 3.2/2.8 kcal mol-1 (PM3/AM1) or 1.6/1.4 for the monomer. These values are
lower than the energies of O...HO interactions obtained using the AM1 or PM3
methods, which tend to be in the range of 3-5 kcal mol-1,
suggesting that [pi]-hydrogen bonds will only form when other structural
factors favour them.
Conclusions. Whilst HO...[pi]-alkene
hydrogen bonding with rH...C < 2.4Å remains a relatively rare
phenomenon, we note that a wide structural diversity appears to be tolerated.
Several of these interactions may directly reflect the reactivity, and we also
anticipate that such bonding will also play a significant role in chiral
recognition and other intermolecular phenomena.[12 ] These
systems should serve a valuable role in evaluating the reliability of both
present modelling methodology such as PM3 and future methodology based on
e.g. a distributed multipole analysis of the charge distribution.
Furthermore, the prospects of specifically designing molecules in which such
interactions are enhanced based on the systems identified here appear
promising.
References.
1. A.W.Baker, A.T.Shulgin,
J. Am. Chem. Soc. 1958, 80, 5358; G.C.Pimental, A.L.McClellan,
"The Hydrogen Bond," Freeman, San Francisco, 1960; Z.Yoshide, E.Osawa, J. Am.
Chem. Soc., 1965, 87, 1467; Z.Yoshide, N.Ishibe, H.Ozoe, J. Am.
Chem. Soc., 1972, 94, 4948; M.D.Joesten, J.L.Schaad, "Hydrogen
Bonding", Dekker, New York, 1974; T.Schaefer, R.Sebastion, T.A.Wildman , Can.
J. Chem., 1979, 57, 3005.
2. A. T. McPhail and G. A.
Sim., Chem. Comm., 1965, 125; A.D.U.Hardy, D.D.MacNicol, J.C.S. Perkin
II, 1976, 1140; S.Ueji, K.Nakatsu, K.Yoshioka, K.Kinoshita, Tet.
Letts., 1982, 23, 1173; G. Valle, M. Crisma, C. Toniolo, N. Sen, M.
Sukumar and P Balaram, J. Chem. Soc., Perkin Trans II, 1988, 393; H.S.Rzepa,
M.L. Webb, A.M.S.Slawin, D.J.Williams, J., Chem. Soc., Chem. Commun.,
1991, 765; S.S.Al-Juaid, A.K.A.Al-Nasr, C.Eaborn, P.B.Hitchcock, J. Chem.
Soc. Chem. Commun., 1991, 1482; J.L.Atwood, F.Hamada, K.D.Robinson, G.W.Orr,
R.L.Vincent, Nature, 1991, 349, 683; G. Valle, M. Crisma, C.
Toniolo, N. Sen, M. Sukumar and P Balaram, J. Chem. Soc., Perkin Trans
II, 1988, 393.
3. M. A. Viswamitra, R. Radhakrishnan, J.
Bandekar and G. R. Desiraju, J. Am. Chem. Soc., 1993, 115,
4868.
4. F. H. Allen, J. E. Daview, J. J. Galloy, O. Johnson, O.
Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G.
Watson, J. Chem. Inf. Comp. Sci., 1991, 31, 187.
5. S. K. Burley and G. A. Petsko, FEBS Lett., 1986, 203, 139.
6. A. J. Stone, Chem. Phys. Lett., 1981, 83, 233; A. J.
Stone and M. Alderton, Mol. Phys., 1985, 56, 1047; S. L. Price, J. S.
Andrews, C. W. Murray and R. D. Amos, J. Am. Chem. Soc., 1992,
114, 8268.
7. H. S. Rzepa and M.Yi, J. Chem. Soc. Perkin
Trans. 2, 1990, 943; I. Juranic, H.S. Rzepa and M. Yi, ibid, 1990,
877. For a discussion of theoretical calculations of C-H...O hydrogen bonding,
see J. J. Dannenburg and L. Turi J. Phys. Chem., 1993, 97,
7899.
8. MOPAC 93.00: J. J. P. Stewart, Fujitsu Limited, Tokyo,
Japan (1993). Available from Quantum Chemistry Program Exchange, University of
Indiana, Bloomington, Indiana.
9. V.E. Steinwender, E. T. G. Lutz,
J. H. van der Maas and J. A. Kanters, Vibrational Spectroscopy, 1993,
4, 217. A survey of known hydrogen bonding interactions to acidic
terminal H-C alkynes was reported by G. R. Desiraju, J. Chem. Soc., Chem
Commun, 1990, 454.
10. M.Geissler, J.Kopf and E.Weiss,
Chem. Ber. 1989, 122, 1395.
11. L. R. Hanton, C.
A. Hunter and D. H. Purvis, J. Chem. Soc. Chem. Commun., 1992, 1134.
12. P. Camilleri, D. Eggleston, H. S. Rzepa and M. Webb. J. Chem.
Soc., Chem. Commun., 1994, 1135
Table. Short O-H...C=C or
C[equivalence]C Contacts From a Quest Search of the Cambridge Structure
Database.
 [a]S. Y.
Lin, Y. Okaya, D. M. Chiou, W.J . Le Noble, Acta Crystallogr., Sect.B,
1982, 38, 1669. OH located. [b]A. Furlani, S.
Licoccia, M. V. Russo, A. C. Villa, C. Guastini, J. Chem. Soc., Dalton
Trans., 1984, 2197. OH located. [c]B. F. Bowden, J. C.
Coll, E. Ditzel, S. J. Mitchell, W.T.Robinson Aust. J. Chem., 1982,
35, 997. Calculated as dimer. H atoms included in refinement. [d]L. A. Paquette, D. R. Andrews, J. P. Springer, J. Org.
Chem., 1983, 48, 1147. [e]L. Quijano, J. S.
Calderon, G. F. Gomez, P. J. Lopez, T. Rios, F. R. Fronczek
Phytochemistry, 1984, 23, 1971. OH located.[f]M. Parvez, K. S. Feldman, B. J. Kosmider, Acta Cryst.,C
(Cr.Str.Comm.), 1987, 43, 1410. OH located. [g]W.
Adam, V. Lucchini, E. M. Peters, K. Peters, L. Pasquato, H. G.von Schnering,
K.Seguchi, H. Walter, B. Will, Chem. Ber., 1989, 122, 133. [h]V. Siemund, H. Irngartinger, C. Sigwart, B. Kissler, R.
Gleiter, Acta Cryst.,C (Cr.Str.Comm.), 1993, 49, 57. OH located.
[i]W. Fenical, G. R. Schulte, J. Finer, J. Clardy,
J.Org.Chem., 1978, 43, 3628. OH located. [j]T.
Wu, M.Wang,T. Jong, A. T. McPhail, D. R. McPhail, K. Lee, J. Nat. Prod.,
1989, 52, 1284. H located. [k]P. C. Coleman, E. D.
Coucourakis, J .A. Pretorius, S.Afr.J.Chem., 1980, 33, 116.
Calculated as dimer. OH located.[l]H. E. Zimmerman, M. J.
Zuraw, J. Am. Chem. Soc., 1989, 111, 7974. OH located. [m]H. J. Cowe, P. J. Cox, R. A. Howie,
J.Cryst.Spectrosc., 1990, 20, 571. [n] As
dimer. [o] As tetramer.
[a]S. Y.
Lin, Y. Okaya, D. M. Chiou, W.J . Le Noble, Acta Crystallogr., Sect.B,
1982, 38, 1669. OH located. [b]A. Furlani, S.
Licoccia, M. V. Russo, A. C. Villa, C. Guastini, J. Chem. Soc., Dalton
Trans., 1984, 2197. OH located. [c]B. F. Bowden, J. C.
Coll, E. Ditzel, S. J. Mitchell, W.T.Robinson Aust. J. Chem., 1982,
35, 997. Calculated as dimer. H atoms included in refinement. [d]L. A. Paquette, D. R. Andrews, J. P. Springer, J. Org.
Chem., 1983, 48, 1147. [e]L. Quijano, J. S.
Calderon, G. F. Gomez, P. J. Lopez, T. Rios, F. R. Fronczek
Phytochemistry, 1984, 23, 1971. OH located.[f]M. Parvez, K. S. Feldman, B. J. Kosmider, Acta Cryst.,C
(Cr.Str.Comm.), 1987, 43, 1410. OH located. [g]W.
Adam, V. Lucchini, E. M. Peters, K. Peters, L. Pasquato, H. G.von Schnering,
K.Seguchi, H. Walter, B. Will, Chem. Ber., 1989, 122, 133. [h]V. Siemund, H. Irngartinger, C. Sigwart, B. Kissler, R.
Gleiter, Acta Cryst.,C (Cr.Str.Comm.), 1993, 49, 57. OH located.
[i]W. Fenical, G. R. Schulte, J. Finer, J. Clardy,
J.Org.Chem., 1978, 43, 3628. OH located. [j]T.
Wu, M.Wang,T. Jong, A. T. McPhail, D. R. McPhail, K. Lee, J. Nat. Prod.,
1989, 52, 1284. H located. [k]P. C. Coleman, E. D.
Coucourakis, J .A. Pretorius, S.Afr.J.Chem., 1980, 33, 116.
Calculated as dimer. OH located.[l]H. E. Zimmerman, M. J.
Zuraw, J. Am. Chem. Soc., 1989, 111, 7974. OH located. [m]H. J. Cowe, P. J. Cox, R. A. Howie,
J.Cryst.Spectrosc., 1990, 20, 571. [n] As
dimer. [o] As tetramer.
Caption to Figure. Three dimensional structures for (a) BETXAZ, (b) DAXWOO, (c) BIWDEQ,
(d) BUTGOM, (e) CUSCIC, (f) FIMNEU, (e) KALHAG, (f) KUMYOG, (i) POITDL, (j)
QQQGTD01, (k) RTRSIN, (l) SEDBIM, (m) VIHGUO. Hydrogen bonds are shown with
dotted lines. These diagrams can be inspected in colour using the electronic
publishing mechanism referred to above.