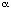 (2-(Dibutylamino)Ethyl)-6-Trifluomethyl-9-Phenanthrene Methanol Hydrochloride(2-(Dibutylamino)Ethyl)-6-Trifluomethyl-9-Phenanthrene Methanol Hydrochloride
(2-(Dibutylamino)Ethyl)-6-Trifluomethyl-9-Phenanthrene Methanol Hydrochloride(2-(Dibutylamino)Ethyl)-6-Trifluomethyl-9-Phenanthrene Methanol Hydrochloride[a]SmithKline Beecham Pharmaceuticals, Coldharbour Road, The Pinnacles, Harlow, Essex, CM19 5AD[ b]SmithKline Beecham Pharmaceuticals, Upper Merion, 709 Swedeland Road, King of Prussia, PA 19406, USA. [c]Department of Chemistry, Imperial College of Science Technology and Medicine, London, SW7 2AY, U.K. E-mail: rzepa@ic.ac.uk. [d]SmithKline Beecham Pharmaceuticals, Old Powder Mills, Leigh, Tonbridge, Kent, TN11 9AN.
 -
face stacking.
facial hydrogen bonding in the solid state
and an asymmetry between the calculated molecular
electrostatic potentials of the two -faces
attributed to antiperiplanar interactions with the CF3
group. Such -asymmetry was also thought to
contribute to chiral resolution of phaclofen on
cyclodextrin HPLC columns[3].
-
face stacking.
facial hydrogen bonding in the solid state
and an asymmetry between the calculated molecular
electrostatic potentials of the two -faces
attributed to antiperiplanar interactions with the CF3
group. Such -asymmetry was also thought to
contribute to chiral resolution of phaclofen on
cyclodextrin HPLC columns[3].
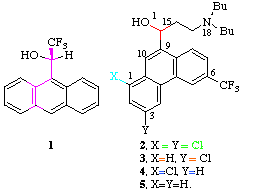 Please click on various parts
of this structure diagram for further explanations
Please click on various parts
of this structure diagram for further explanations
As part of our effort to
enhance the ability to predict structural requirements
necessary for chiral resolution, we have compared the
chromatographic behaviour of the anti-malarial compound halofantrine [4]
(2, R=(CH2)2NBu2) with two closely related
monochloro derivatives 3 and 4 using a
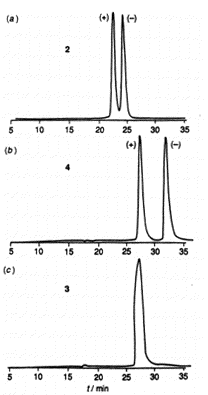
Pirkle type chiral stationary phase. Figure 1 shows that whereas the resolution of the
enantiomers of 4 (separation factor 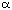 1.38) is actually increased compared to that of 2
( 1.30) the enantiomers of 3 co-elute.
The retention time of 3 is the same as that of the
faster eluting enantiomer (+)-4, which suggests
the longer retention time of (-)-4 is largely due
to chiral recognition. The 1,3-H isomer (5) also
resolves on the column, but less well than 4 or
2 1.20) indicating that the 3-Cl group
also plays a significant role in suppressing chiral
recognition. We felt it imperative therefore to establish
the reasons for the differing chiral behaviour of these
systems.
1.38) is actually increased compared to that of 2
( 1.30) the enantiomers of 3 co-elute.
The retention time of 3 is the same as that of the
faster eluting enantiomer (+)-4, which suggests
the longer retention time of (-)-4 is largely due
to chiral recognition. The 1,3-H isomer (5) also
resolves on the column, but less well than 4 or
2 1.20) indicating that the 3-Cl group
also plays a significant role in suppressing chiral
recognition. We felt it imperative therefore to establish
the reasons for the differing chiral behaviour of these
systems.
By analogy with 1, we initially
sought an explanation for the differing behaviour of
2 and 3 in the properties of the
-faces, via calculation of the molecular
electrostatic potentials (MEPs).[2]
Since we have observed that this property might be
sensitive to the orientation of the OH group, the x-ray
structure of the
crystalline hydrochloride derivative of 3 was
obtained; that for 2 has been previously
measured.[5] These structures revealed
strong hydrogen bonds to form in both compounds from the
OH group to the halide counter-anion. However, PM3
calculations[6] based on the x-ray
derived OH orientation revealed no significant
differences in the MEPs of 2 and 3 (R=H).
In both cases the most negative region resides over the A
rather than C ring, consistent with the A-C ring
complementary - stacking observed[5] for 2.
The crystal
structure of a solvated form of the hydrochloride salt of
3 shows two crystallographically independent
molecules which comprise the asymmetric unit, referred to
as molecules A and B, and one molecule of isopropanol.
The two molecules are virtually identical in their
molecular conformation. The C10-C9-C15-O1 torsion angles
(near 24[o]), which define the
rotameric orientation of the hydroxyl group relative to
the central ring, are remarkably small. The hydroxyl
hydrogen in both molecules lies in an orientation which
is perpendicular to the plane of the phenanthrene ring,
on the side of this ring opposite to the
dibutylaminopropyl substituent at C17. This orientation
is different from those observed for the hydroxyl
hydrogens in the structure of halofantrine[5]. In that molecule, the hydroxyl
hydrogens point toward the dibutylaminopropyl substituent
and are more nearly parallel to the plane of the
phenanthrene ring.The packing mode in 2 is
strikingly absent in the lattice of racemic 3,
where the alkyl side chains fold over the -faces,
and the rings instead adopt a complementary edge-to-edge
dimeric interaction between pairs of enantiomers (Figure
2). This interaction involves contacts between an
apparently electron deficient aromatic C1-H and a lone
pair of the oxygen atom from a second molecule, slightly
staggered to avoid contact between the two C10-H atoms
(Figure 2). Although weak
aromatic C-H hydrogen bonding has often been observed,[7] the H...O distance of 2.3Å in
3 is relatively short and in this case can be
attributed to the acidity of the 1-H hydrogen induced by
a combination of a "W" interaction with the 3-Cl
substituent and a longer range seven bond antiperiplanar
(app) interaction along the 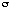 -framework
with the CF3 substituent. A search of the Cambridge
structural database[8] revealed that
although similar hydrogen bonding is induced
by m-chloro aromatic substition,[9] no examples of longer range CF3
induced interactions appear to have been reported. []
-framework
with the CF3 substituent. A search of the Cambridge
structural database[8] revealed that
although similar hydrogen bonding is induced
by m-chloro aromatic substition,[9] no examples of longer range CF3
induced interactions appear to have been reported. []
PM3 calculations[6] for the model system where the dibutylamino group (R) is
replaced by R=H, reveal the charge on C1-H (0.1039 for
5, R=H, 6-CH3) is approximately equally increased
by the 6-CF3 and 3-Cl groups (0.1060 for 5, R=H;
0.1067 for 3, R=H, 6-CH3; 0.1087 for 3,
R=H), suggesting both groups are necessary for hydrogen
bonding at this position to influence chiral
resolution.[#] This is supported by
our observation that 5 can be resolved, unlike
3. The replacement of C1-H with C1-Cl as in
2 or 4 would also inhibit such an
interaction, favouring instead the - stacking
mode which we believe is partially responsible for chiral
recognition. We also note the substantial analogy between
3 and the cyclic dimeric structure (6) of
thiamine picrate[10], where an even
stronger hydrogen bond (rH...O 2.21Å) to the acidic
2-H ring hydrogen is formed.

An attempt was made to study the
interactions responsible for the HPLC results using
solution [1]H nmr. Initially, the free
bases of 2 and 3 were studied in the
presence of the methyl ester of dinitrobenzoyl leucine.
Even at -40deg.C, the observed [1]H
shifts and nonequivalences were too small
(<0.01-0.05ppm) to make worthwhile comparisons, a
reflection of the small differences in binding free
energy between the diastereomeric complexes (Figure 1).
However, the use of S(+) 1 (36 mM) as the chiral
solvating agent for the free base forms of 2 and
3 (20 mM in CDCl3) revealed a significant (0.24
ppm) upfield shift of the 10-H signal of 2 with an
induced nonequivalence of 0.02 ppm, with similar values
for 4 (0.15/0.02ppm). In contrast the values for
3 are 0.04/0.01 ppm. The H-8 signals for all three
complexes show comparable upfield shifts of ~0.25 ppm and
induced nonequivalence of ~0.1 ppm. We interpret this as
indicating significant -stacking character in the
diastereomeric complexes between 1 and
2, whereas additional edge-to-edge
interactions to 3 would induce downfield shifts at
H-10 which largely offset upfield shifts due to
-stacking. This supports our hypothesis that the
chiral HPLC recognition is associated with -
stacking interactions in 2, whereas stronger
edge-to-edge, and apparently achiral, hydrogen bonding
interactions eliminate the chiral recognition to
3.
On the basis of our results, we predict
that the shorter range double app interaction in
the model compound 7 should also result in C-H...O
hydrogen bonding to the 9-H hydrogen. This prediction is
supported by two types of PM3 calculation. The charge on
9-H (0.1105, 7; R=H) vs the unfluorinated
analogue (0.1070, 7, R=H, 4,6-CH3) is increased
compared with 3 (R=H). Secondly, the calculated
enthalpy difference of +7.0 kcal mol[-1
]between 3 (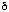 H -151.1 kcal mol[-1], R=H) and its 1-anion (H
-144.1 kcal mol[-1]) is reduced to
+0.3 for (7, R=H). The corresponding values for
5; R=H, 6-CH3, 5; R=H, and 3, R=H,
6-CH3 are 18.9, 12.3 and 13.8 kcal mol-1
]respectively, values which also support our
contention that the electronic effects of the 3-Cl and
6-CF3 groups in 3 are approximately equal.
H -151.1 kcal mol[-1], R=H) and its 1-anion (H
-144.1 kcal mol[-1]) is reduced to
+0.3 for (7, R=H). The corresponding values for
5; R=H, 6-CH3, 5; R=H, and 3, R=H,
6-CH3 are 18.9, 12.3 and 13.8 kcal mol-1
]respectively, values which also support our
contention that the electronic effects of the 3-Cl and
6-CF3 groups in 3 are approximately equal.
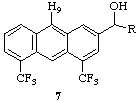
Crystal Data for
3: C26H31ClF3NO*HCl*1/2(C3H7OH), crystallized from
isopropanol, Mr = 531.06, colorless, rectangular plate,
0.80 x 0.30 x 0.10 mm, triclinic, P-1, T = 223K, a =
8.561(6)Å, b = 18.180(6)Å, c =
19.286(3)Å, = 75.41(3)[o], 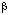 = 77.28(4)[o],[ ]
= 77.28(4)[o],[ ]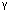 =
76.26(4)o, Volume =
2780.4(12)Å[3], Z = 4,
=
76.26(4)o, Volume =
2780.4(12)Å[3], Z = 4, (calc) = 1.269 g.cm[-3], u = 2.782
cm[-1], F(000) = 1174, Enraf-Nonius
CAD4 diffractometer, MoK- ([[lambda]] =
0.71073Å), lattice parameters determined from the
setting angles of 25 high order reflections, variable
speed [[omega]]-2[[theta]] scans for data collection in
the range: 2deg. < 2[[theta]] <
50deg., indices: 0 < h < 10, -21
< k < 21, -22 < l
< 22, 9937 reflections, 9217 unique (Rint =
0.053), data corrected for Lorentz and polarization
factors, 3040 data I > 3.0(I), refined
in blocks of 298 variables, weights = 1/[2](Fo) with [2](Fo) = 1/[[2](Io) + (0.04*Io)[2]], R = 0.078, Rw = 0.093, goodness of
fit = 1.897, final difference map residual density
between +/- 0.885 eÅ[-3], final
/ = 0.01, neutral atom scattering
factors. The structure was solved by direct methods using
the SHELXS program series. Atomic positions were
initially refined with isotropic temperature factors and
subsequently with anisotropic displacement parameters.
The function minimized was w(|Fo|-|Fc|)[2]. The trifluoromethyl group fluorine
atoms show high isotropic thermal values which may be
indicative of rotational disorder, but a chemically
sensible model could not be refined for the data. The
solvent molecule is disordered; it was treated
isotropically at half-occupancy in a "best-fit mode".
Positions for hydrogen atoms attached to carbons were
assigned based on geometrical considerations (C-H distances 1.0-1.1A) except that
hydrogen atoms were omitted from the disordered solvent
molecule. Positions for hydrogens attached to heteroatoms
were calculated based on consideration of the putative
hydrogen bonding scheme (N-H, O-H distances 1.0-1.1A) and were included in the final
refinement stages as fixed contributions with isotropic
temperature factors assigned 1.3(Beq) of the attached
atom. Programs in the SDP package were used for data
reduction and refinement.
(calc) = 1.269 g.cm[-3], u = 2.782
cm[-1], F(000) = 1174, Enraf-Nonius
CAD4 diffractometer, MoK- ([[lambda]] =
0.71073Å), lattice parameters determined from the
setting angles of 25 high order reflections, variable
speed [[omega]]-2[[theta]] scans for data collection in
the range: 2deg. < 2[[theta]] <
50deg., indices: 0 < h < 10, -21
< k < 21, -22 < l
< 22, 9937 reflections, 9217 unique (Rint =
0.053), data corrected for Lorentz and polarization
factors, 3040 data I > 3.0(I), refined
in blocks of 298 variables, weights = 1/[2](Fo) with [2](Fo) = 1/[[2](Io) + (0.04*Io)[2]], R = 0.078, Rw = 0.093, goodness of
fit = 1.897, final difference map residual density
between +/- 0.885 eÅ[-3], final
/ = 0.01, neutral atom scattering
factors. The structure was solved by direct methods using
the SHELXS program series. Atomic positions were
initially refined with isotropic temperature factors and
subsequently with anisotropic displacement parameters.
The function minimized was w(|Fo|-|Fc|)[2]. The trifluoromethyl group fluorine
atoms show high isotropic thermal values which may be
indicative of rotational disorder, but a chemically
sensible model could not be refined for the data. The
solvent molecule is disordered; it was treated
isotropically at half-occupancy in a "best-fit mode".
Positions for hydrogen atoms attached to carbons were
assigned based on geometrical considerations (C-H distances 1.0-1.1A) except that
hydrogen atoms were omitted from the disordered solvent
molecule. Positions for hydrogens attached to heteroatoms
were calculated based on consideration of the putative
hydrogen bonding scheme (N-H, O-H distances 1.0-1.1A) and were included in the final
refinement stages as fixed contributions with isotropic
temperature factors assigned 1.3(Beq) of the attached
atom. Programs in the SDP package were used for data
reduction and refinement.
Computer readable files for Apple Macintosh and Microsoft Windows systems in Quicktime(TM) and MPEG video animation format illustrating the three dimensional properties of 2 and 3 and bond length and angle information are available for general access from the chemistry Gopher+ server gopher.ch.ic.ac.uk. These files will reside in the Royal_Society_of_Chemistry/Chemical_Communications/3_03989G directory for a period of at least two years from the publication of this paper. A description of how to visualise such material, together with appropriate programs is available from the same source. The material can also be obtained by connecting to the following world-wide-webb service: http://www.ch.ic.ac.uk/rzepa/RSC/CC/3_03989G.html.
[#] On this basis we predict that the HPLC behaviour of (3, 6-CH3) should be similar to that of 5. We thank a referee for this suggestion. Unfortunately, neither this material nor compound 7 is available to us. []
References.
1. W. H. Pirkle, D. J. Hoover, Top. Steriochem., 1982, 13, 263; G. R. Wiesman, Assymetric Synthesis, Ed. J. D. Morrison, Vol. 1, Academic Press, London, 1983; K. B. Lipkowitz, D. A. Dementer, R. Zegarra, R. Larter, T. Darden, J. Am. Chem. Soc., 1988, 110, 3446; I. W. Wainer, T. D. Doyle, J. Chromatography, 1984, 284, 117; D. M. McDaniel, B. G. Snider, J. Chromatography, 1987, 404, 122; W. H. Pirkle, J. E. McCune, J. Chromatography, 1989, 469, 67.
2. H.S. Rzepa, M. L. Webb, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 765.
3. P. Camilleri, H. S. Rzepa and S. M. Green, J. Chem. Soc., Chem. Commun, 1992, 1122.
4. Halofantrine, is an antimalerial drug marketed by Smithkline Beecham,"Merk Index" 1989, 11th ed. 4504; P. Camilleri, C. Dyke and F. Hossner, J. Chromatogr., 1989, 477, 471.
5. The structure of 2 has been previously reported, but the R factor was 18.3%; J. M. Karle and I. L. Karle, Acta Cryst.,C, 1989, 45, 1248. We have redetermined this structure and refined it to an agreement of 15.6%. Data are available on request from the authors.
6. J. J. P. Stewart, J. Comp. Chem., 1989, 10, 209, 221 implemented in MOPAC-93 from Fujitsu, Tokyo, Japan, and available from Quantum Chemistry Program Exchange, University of Indiana, Bloomington, Indiana.
7. R. D. Green "Hydrogen Bonding by C-H Groups", Wiley Interscience, New York, 1974. R. Taylor, O. Kennard, J. Am. Chem. Soc., 1982, 104, 5063. The shortest aromatic C-H...O interaction quoted in this paper is found in 1,3,5-trinitrobenzene at 2.246Å. See also T. Steiner and F.Saenger J. Am. Chem. Soc., 1992, 114, 10146. Hydrogen bonding is also known to acidic terminal H-C alkynes ; G. R. Desiraju, J. Chem. Soc., Chem Commun, 1990, 454.
8. F. H. Allen, J. E. Daview, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Comp. Sci., 1991, 31, 187.
9. X.Li, M.S.Lah ,V.L.Pecoraro, Acta Cryst.,C, 1989, 45, 1517.
10. M.-J. Kim, I.-H. Suh, K.Aoki and H.Yamazaki Acta Cryst.C, (Cr.Str.Comm.), 1988, 44, 725. See also C. L. MacLaurin and M.F.Richardson, ibid, 1983, 39, 854.
Captions to Figures.
Figure 1. Chromatographic behaviour of (a) 2,
(b) 4, (c) 3. HPLC conditions: chiral
column, 250mm x 4.9mm ID; stationary phase,
L-N-(3,5-dinitrobenzoyl) leucine covalently bound to
3-aminopropyl silican support (particle size 5 um);
mobile phase, n-hexane/chloroform/propan-2-ol (90:5:5
v/v/v) containing 1% triethylamine; flow rate 0.4 ml
min[-1] at -5C; detection, uv at
320nm. The value of the separation factor can
be calculated from the chromatographic data, and using
the equation G = RT ln,
results in free energy differences of 585, 718 and 0 J
mol[-1] for 2 4 and 3
respectively.
Figure 2. Crystal structure of 3 showing the crystal stacking from two views (a) and (b), with the C-H...O interactions indicated as dotted green lines. There is an extensive network of conventional hydrogen bonding in the crystal structure of 3 with all available donors participating. Metrical details are: O1A----Cl2A = 3.02(1)Å, HO1A---Cl2A = 2.0Å, angle at hydrogen = 180[o]; O1B----Cl2B = 3.045(7)Å, HO1B---Cl2B = 2.0Å, angle = 178[o]; N18A----Cl2A = 3.052(9)Å, H13------Cl2A = 2.0Å, angle = 170[o];[ ] N18B----Cl2B = 3.06(1)Å, H44------CL2B = 2.1, angle = 168[o]; O100-----Cl2A = 3.46(2)Å, H100-----Cl2A = 2.3Å, angle = 180[o]. Metrical details for the C-H..O interactions are: C3A----O1B = 3.29(1)Å, H1---O1B = 2.3Å, angle = 159[o]; C3B----O1A = 3.27(1)Å, H33----O1A = 2.3Å, angle = 155[o].
{kind=link}
{kind=link}