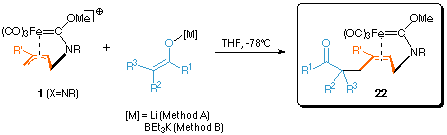
| Lithium enolates, potassium enoxy borates, cuprates and phosphanes attack at the allyl terminus of 1 (X=NR) to form alkene carbene complexes 22[5] which are sufficiently stable to be isolated (in contrast to their oxa-analogues, see Path B).
| 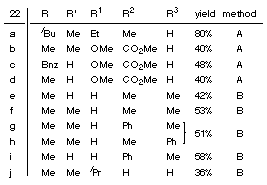 |
| X-ray structure of 22a: |
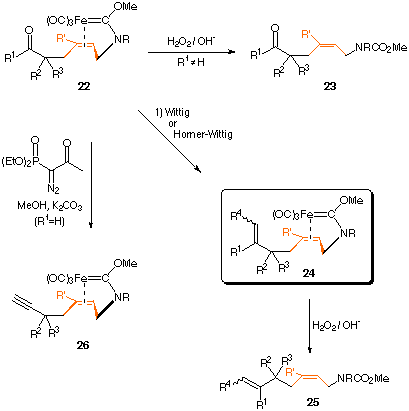
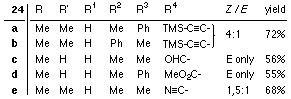
 | Demetalation of 22 with H2O2/OH- leads to carbamates 23. |
Prior to decomplexation of the organic ligand, various transformations of the carbonyl group
of 22 can be performed (compare to path b).
| Wittig or Horner-Wittig olefination of 22 gives carbene complexes of type 24 optionally with additional functionalities like alkynyl-, aldehyde-, keto-, ester- or nitril-groups. Demetalation with H2O2/OH- leaves 25. |
| Alternatively, the carbonyl group in "formyl-terminated" complexes 22 can be directly converted to a 1-alkyne unit by reaction with 1-diazo-2-oxo-propyldimethylphosphonate under slightly basic conditions. |
Experimental data
1. Synthesis of 22a-d (general procedure method A):
2. Synthesis of 22e-k (general procedure method B):
3. Synthesis of 24d
4. Synthesis of 24a-c*,f (general procedure):
[5] J. Böhmer, W. Förtsch, F. Hampel, R. Schobert Chem. Ber. 1996, 129, 427.
n-BuLi (0.48 ml of a 2.5 M solution in hexane; 1.2 mmol) was added to a solution of N-isopropylcyclohexylamine (170 mg; 1.2 mmol) in THF
(5 ml) at 0 °C. The mixture was stirred for 30 min, then cooled to -78 °C and treated with the carbonyl compound (1.2 mmol). After 1 h the resulting solution was transferred by means of a cannula to a slurry of 1c (354 mg; 1.0 mmol) in THF, chilled to
-78 °C (or -40 °C in the case of cyclic ketones). Instantaneous deepening of the yellow shade indicated a swift reaction. Any volatile components were evaporated after another hour and the residue was repeatedly extracted with ether/hexane (2:1). The crude product thus obtained was subsequently purified by CC (silica; ether/petroleum ether, 1:1).
(4Z)-Tricarbonyl[(4-5-eta2)-2-aza-2-benzyl-7-bis(methoxycarbonyl)-1-methoxy-4-hepten-1-ylidene]iron(0) (22c)
IR (neat): 3015, 2940, 2860, 2005, 1940, 1910, 1745, 1730 , 1520, 1450,1430, 1220 cm-1. - 1H NMR (CDCl3, 400 MHz): 2.03 [ddd, 2J (6-Ha/6-Hb) = 12.00 Hz, 3J (6-Ha/7-H) = 4.80 Hz, 3J (6-Ha/5-H) = 10.70 Hz, 1H, 6-Ha], 2.30 [ddd, 3J (6-Ha/6-Hb) = 12.00 Hz, 3J (6-Hb/5-H) = 6.00 Hz, 3J (6-Hb/7-H) = 5.60 Hz, 1H, 6-Hb], 2.50 [dd, 2J (3-Ha/3-Hb) = 13.70 Hz, 3J (4-H/3-Hb) = 4.90 Hz, 1H, 3-Hb], 3.43 [m, 1H, 3-Ha], 3.51 [mc, 1H, 7-H], 3.73 [s, 6H, CO2CH3], 3.81 [mc, 3H, 3-Ha, 4-H, 5-H], 4.20 [s, 3H, CO2CH3], 4.28 [d, 2J = 14.29 Hz,1H, N-CH2], 4.71 [d, 2J = 14.29 Hz, 1H, N-CH2], 7.13 [m, 2H, aromat. H], 7.31 [m, 3H, aromat. H]. - 13C NMR (CDCl3, 100.5 MHz): d = 31.02 (C-3), 46.78 (C-4 or C-5), 49.40
(C-6), 50.65 (C-4 or C-5), 52.40 (COCH3), 53.86 (C-7), 54.34 (N-CH2), 62.81(COCH3), 127.67, 127.83, 128.77, 134.79 (aromat. C), 169.40/169.88 (CO2CH3), 215.78 (Fe=CO), 238.97 (C-1).Ę- MS 70 (eV); m/z (%): 455 (11) [M+ - CO], 389 (100) [M+ - 3CO], 152 (74) [{389} - C6H5CH2+], 278 (40), 198 (66), 148 (29), 91 (83) [C6H5CH2+]. - C21H23FeNO8 (473.3): calcd. C 53.30,
H 4.90, N 2.96; found C 53.24, H 4.90, N 3.08.
Solutions of the potassium enolates were prepared by dropwise addition of the respective carbonyl compound (0.78 mmol) to a suspension of potassium hydride (38 mg, 0.94 mmol) in THF (3 mL) at room temperature. After stirring vigorously for 10 min the solution was separated from excess potassium hydride by filtration over a glass sinter funnel. The resulting clear colourless to yellow filtrate was treated with triethylborane (0.98 mL of a
1 M solution in THF, 0.98 mmol) to give the corresponding enoxyborate. This solution was slowly transferred by means of cannula to a slurry of 1c (200 mg, 0.57 mmol) in THF (5 mL), kept at -78 °C. After one hour the reaction mixture was warmed to ambient temperature (25 °C) and any volatile components were evaporated in vacuo. The residue thus obtained was redissolved in the minimum amount of ether/pentane (1:1) and subsequently purified by column chromatography.
(4Z)-Tricarbonyl[(4-5-eta2)-2-aza-1-methoxy-2,7,7-trimethyl-4-octen-1-yliden-8-al]iron (0)(22e)
IR (neat) :2960, 2920, 2830, 2705, 2015, 1910, 1725, 1545, 1460 cm-1. -
1H NMR (C6D6): 0.85 and 0.86 [s each, 6H, 7-CH3], 1.19 [dd, 3J (3-Hex / 4-H) = 10.7 Hz, 2J (3-Hex / 3-Hen) = 14.5 Hz,1H, 3-Hex], 2.01 [s, 3H, N-CH3], 2.14 [dd, 3J (3-Hen / 4-H) = 3.0 Hz, 2J (3-Hen / 3-Hex) = 14.5 Hz, 1H, 3-Hen], 2.58-2.64 [m, 1H, 4-H], 3.20 [d, 3J (6-H / 5-H) = 3.9 Hz, 2H, 6-H], 3.27-3.31 [m, 1H, 5-H], 3.70 [s, 3H, O-CH3]. -
13C NMR (C6D6): 21.3, 21.6 (7-CH3), 31.3 (N-CH3), 39.9 (C-3), 47.7 (C-9), 48.4, 49.2 (C-4, C-5), 56.6 (C-6), 62.4 (O-CH3), 205.3 (C-8), 216.7 (Fe=CO), 238.4 (C-1). -
MS ( 70 eV) : m/e (%) = 337 (1) [M+], 309 (23) [M+-(CO)], 281 (6) [M+-2(CO)], 253 (87) [M+-3(CO)], 183 (100) [Fe(CO)3(COMe)+], 142 (37), 102 (58).
24d was synthesized via Wittig-reaction with Methoxy-carbonyl triphenylphosphorane in pentane in the presence of silica gel [7].
A suspension of KH in THF was treated with the phosphonate under vigorous stirring at room temperature. After 5 min the solution was cooled to -78°C and slowly transferred by means of cannula to a solution of 22 (200 mg, 0.57 mmol) in THF (5 mL), kept at -78 °C. After one hour at this temperature the solution was allowed to warm to room temperature and quenched with saturated NH4Cl solution. After removal of the solvent the crude product was subjected to column chromatography (ether/petroleum ether 1:5).
(* For the synthesis of 24b diethyl cyclohexylvinylamino phosphonate was used. The imine formed primarily hydrolyzes during chromatography to leave the aldehyde.)
(4Z)(8E)-Tricarbonyl[(4-5-eta2)-2-aza-9-cyano-1-methoxy-2,5,7,7-trimethyl-4,8-nonadien-1-ylidene]iron (0)
IR (Film) : 3060, 2960, 2935, 2850, 2220, 2010, 1940, 1700, 1550 cm-1. -
1H NMR (C6D6): 0.60 and 0.64 [s each, 6H, 7-CH3], 1.30 [m, 1H, 3-Hex], 1.72 [s, 3H, 5-CH3], 1.87 [dd, 3J (3-Hen/4-H) = 2.1 Hz, 2J (3-Hen/3-Hex) = 14.2 Hz, 1H, 3-Hen], 2.07 [s, 3H, NCH3], 2.51-2.54 [m, 1H, 4-H], 3.00 [d, 2J (6-Ha/6-Hb) = 13.7 Hz, 1H, 6-Ha], 3.13 [d, 2J (6-Ha/6-Hb) = 13.7 Hz, 1H, 6-Hb], 3.71 [s, 3H, O-CH3], 4.63 [d, 3J (8-H/9-H) = 16.6 Hz, 1H, 9-H], 6.19 [d, 3J (8-H/9-H) = 16.6 Hz, 1H, 8-H]. -
13C NMR (C6D6): 24.7, 26.0 (7-CH3), 29.9 (5-CH3), 31.4 (N-CH3), 40.4 (C-7), 45.3 (C-3), 54.4 (C-4), 61.3 (C-6), 62.3 (O-CH3), 63.0 (C-5), 97.4 (C-9), 117.8 (C-10), 163.5 (C-8), 217.0 (Fe=CO), 239.0 (C-1). -
MS ( 70 eV) : m/e (%) = 318 (7) [M+], 262 (12) [M+-2(CO)], 234 (100) [M+-3(CO)], 162 (44).
(4Z)(8Z)-Tricarbonyl[(4-5-eta2)-2-aza-9-cyano-1-methoxy-2,5,7,7-trimethyl-4,8-nonadien-1-ylidene]iron (0)
1H NMR (C6D6): 1.00 and 1.06 [s each, 6H, 7-CH3], 1.34 [dd, 3J (3-Hen/4-H) = 11.0 Hz, 2J (3-Hen/3-Hex) = 14.3 Hz, 1H, 3-Hex], 1.79 [s, 3H, 5-CH3], 2.00 [dd, 3J (3-Hen/4-H) = 2.2 Hz, 2J (3-Hen/3-Hex) = 14.3 Hz, 1H, 3-Hen], 2.24 [s, 3H, NCH3], 2.63-2.65 [m, 1H, 4-H], 3.16 [d, 2J (6-Ha/6-Hb) = 13.8 Hz, 1H, 6-Ha], 3.26 [d, 2J (6-Ha/6-Hb) = 13.8 Hz, 1H, 6-Hb], 3.72 [s, 3H, O-CH3], 4.56 [d, 3J (8-H/9-H) = 12.6 Hz, 1H, 9-H], 5.65 [d, 3J (8-H/9-H) = 12.6 Hz, 1H, 8-H].
13C NMR (C6D6): 26.5, 26.8 (7-CH3), 30.0 (5-CH3), 31.7 (N-CH3), 40.8 (C-7), 45.6 (C-3), 55.0 (C-4), 61.3 (C-6), 62.3 (O-CH3), 63.6 (C-5), 96.7 (C-9), 116.8 (C-10), 162.5 (C-8), 218.0 (Fe=CO), 238.5 (C-1).
[7] V. J. Patil, U. Mävers Tetrahedron Letters 1996,37, 1281.


