| [Related articles/posters: 086 054 122 ] |
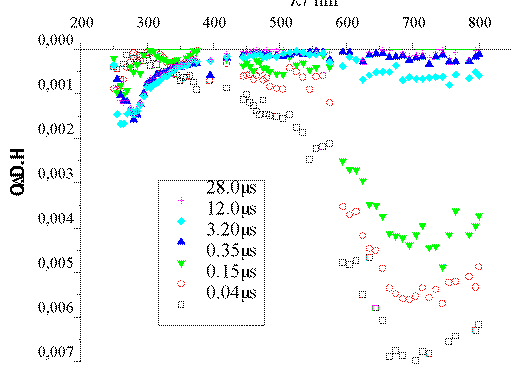
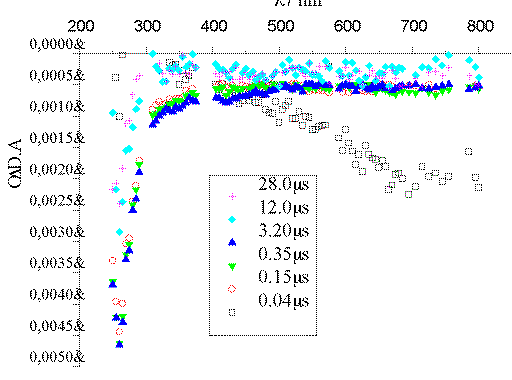
The intense band centered at 720 nm that appears in the Ar-saturated
experiment (Figure 1) corresponds to e-aq.[13] In the presence of O2 (Figure 2), an excellent
e-aq scavenger, such band disappears within 100 ns. The
scavenging capacity of O2 is explained by the formation of
O2*- through the process O2 +
e-aq -> O2*-. The data available
are not enough to identify the transient species absorbing at ca. 270
nm.
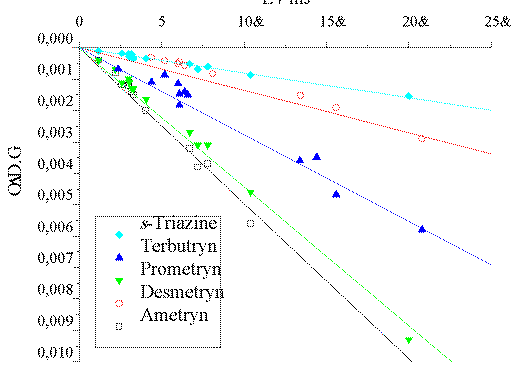
For all the studied compounds the 193 nm photoionization is a monophotonic
process, as determined from [Delta]O.D. (e-aq) vs.
(E / mJ) plots (shown in Figure 3). The photoionization quantum yields
(fPI) were also obtained by comparing the yield of
e-aq in the LFP of the different triazines with that of
e-aq for NaCl actinometry solutions of the same optical
density as the substrates at the excitation wavelength
(fPI(Cl-)=0.41±0.08).? The obtained fPI values are compiled
in Table 1. The very low values obtained suggest the existence of many
effective decay pathways other than electron photoejection for the excited
states generated upon photolysis to relax. It also appears as if the presence
of bulky electron donating groups increases the ease of photoionization, which
would be in agreement with a stabilization of the so-formed radical cation.
Model |
fPI |
s-Triazine |
0.005 |
Herbicides |
fPI |
Ametryn |
0.071 |
Desmetryn |
0.070 |
Prometryn |
0.129 |
Terbutryn |
0.129 |
In all cases the electron photoejection and electron hydration processes took
place within the laser pulse, i.e.: in less than 20 ns. This is in
agreement with previous observations of such processes taking place in less
than 27 ps,[14] and would, in practice, mean
that no ion-pair recombination takes place after the laser pulse.[15]
The results presented here allow us to predict that the photoionization
threshold of triazine derivatives must be around 9.9 eV, of which 6.4 eV are
provided by the exciting 193 nm photons and 3.5 eV come from the hydration of
the photoionization products.[16]
On the basis of the experimental evidences available, the general mechanism
proposed in Scheme 1 can be put forward for the far-UV induced photodegradation
of triazines:
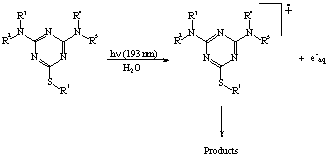
From an environmental point of view, it is remarkable the fact that for all
the studied triazines the only photo-initiated processes were observed when
exciting with 193 nm light, which is not contained in the solar spectral
irradiance.[17] This, in practice, means that
s-triazine based herbicides do not undergo photodegradation upon
exposition to sunlight.
Further research is in progress on photosensitization based degradation of
s-triazines, as well as on the applications of radiation chemistry for the
degradation of such compounds.
Conclusion.
Triazine-based herbicides are not photodegraded by sunlight. Instead, these
compounds can be photodegraded (with low yields) by the action of far-UV
radiation.
Acknowledgments.
M.I.F. acknowledges a Deutscher Akademischer Austauschdienst (DAAD, Germany)
grant. MCL thanks the EU for a Training and Mobility of Researchers contract to
work in the Max-Planck Institut für Strahlenchemie (Mülheim an der
Ruhr, Germany), and for supporting a series of visits to the Paterson Institute
for Cancer Research Free Radical Research Facility (Manchester, U.K.) within
the Access to Large Scale Facilities activity. Thanks are also due to the
Universidade da Coruña for different leaves of absence to MCL.
References.
[1] Worthing, R.C.; The pesticide
manual, 9th ed., British Crop Protection Council, Surrey (U.K.),
1991.
[2] National Research Council. Regulating
Pesticides in Food. National Academy Press, Washington, D.C., 1987.
[3] Ashton, F.M.; Crafts, A.S. Mode of Action
of Herbicides. Wiley, N.Y, 1973.
[4] (a) Spear, R.; Chap. 6 in: Handbook of
Pesticide Toxicology. Hayes, W.J.; Laws, E.R. (Eds.), vol. 1, Academic
Press, Inc., San Diego, 1991. (b) Miles, C.J.; Chap. 5 in Pesticide
Transformation Products. Fate and Significance in the Environment.
Somasundaram, L.; Coats, J.R. (Eds.), ACS Symp. Ser. 459, ACS, 1991; (c)
Belluck, D.A.; Benjamin, S.L.; Dawson, T.; Chap 18 in Pesticide
Transformation Products. Fate and Significance in the Environment.
Somasundaram, L.; Coats, J.R. (Eds.), ACS Symp. Ser. 459, ACS, 1991; (d)
Aherne, G.W.; Chap. 4 in Chemistry, Agriculture and the Environment.
Richardson, M.L. (Ed.), RSC, London, 1991. (e) Funari, E.; Bottoni, P.;
Giuliano, G.; Chap. 14 in in Chemistry, Agriculture and the Environment.
Richardson, M.L. (Ed.), RSC, London, 1991.
[5] (a) Galassi, S.; Guzzella, L.
Acqua-Aria, 1990, 3, 231. (b) Hormann, W.D.; Tournayre,
H.; Egli, H. Pest. Monit. J., 1979, 13, 128.
[6] Grover, R.; Cessna, A.J. (Eds.)
Environmental Chemistry of Herbicides. Vol. II, CRC Press, Boca Raton
(Florida), 1991.
[7] (a) Hodgson, E.; Lefi, P.E. Enviroment
Health Perspectives, 1996, 104, 97. (b) Drevenkar, V.;
Fingler, S.; Fröbe, Z. Chemical Safety International Reference
Manual, pp.297-310(1994).
[8] Hörmann, W.D.; Tourmayre, J.C.; Egli,
H. Pesticides Monitoring Journal, 1979, 13, 128.
[9] (a) Erickson, L.E.; Lee, K.H. CRC
Critical Rev. in Env. Control, 1989, 19, 1. (b) Fairhead,
A.P. J. Inst. Water Env. Manag., 1994, 8, 399. (c)
Hapeman, C.J. ACS Symp. Ser., 1994, 554, 223. (d) Mascolo,
G.; López, A.; Földényi, R.; Passino, R.; Tiravanti, G.
Environ. Sci. Technol., 1995, 29, 2987. (e) López,
A.; Mascolo, G.; Földenyi, R.; Tiravanti, G.; Santori, M. Water
Supply, 1995, 13, 265.
[10] (a) Schmidt, S.; Mattusch, J.; Werner, G.
Z. Chem., 1989, 29, 239. (b) ernák, O.;
ernáková, M. Water Supply, 1992, 171. (c) Muszkat,
L.; Geigelson, L.; Bir, L.; Muszkat, K.A. Chemosphere, 1998,
36, 1485.
[11] PI(Cl-)=0.41.
Iwata, A.; Nakashima, N.; Kusaba, M.; Izawa, Y.; Yamanaka, C. Chem. Phys.
Lett. 1993, 207, 137.
[12] Hug, G.L. Natl. Stand. Ref. Data
Ser. 1981, 69, 1
[13] Jou, F.Y.; Freeman, G.R. J. Phys.
Chem. 1977, 81, 909.
[14] Mialocq, J.C.; Amouyal, E.; Bernas, A.;
Grand, D. J. Phys. Chem. 1982, 86, 3173.
[15] M. Canle L., J.A. Santaballa, S.
Steenken. Submitted to publication (1998).
[16] (a) De Violet, P.F. Rev. Chem.
Interm. 1981, 4, 121. (b) Braun, M.; Fan, J.Y.; Fuss, W.;
Kompa, K.L.; Müller, G.; Schmid, W.E. Methods in Laser
Spectroscopy; Prior, Z.; Ben-Reuven, A.; Rosenbluh, M., Eds., Plenum Press,
N.Y. (U.S.A.), 1986.
[17] Murov, S.L.; Carmichael, I.; Hug, G.L.
Handbook of Photochemistry. 2nd de. Marcell Dekker, Inc.,
N.Y. (U.S.A.), 1993.