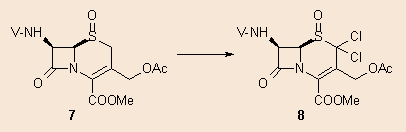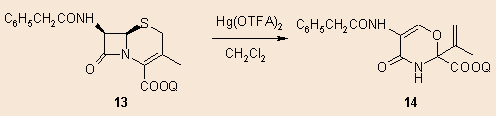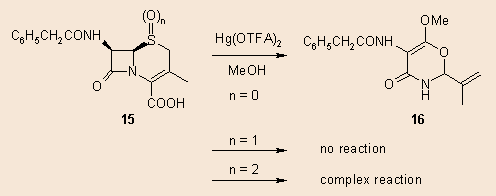aResearch Group for Antibiotics, L. Kossuth University, POB 36, H-4010 Debrecen, Hungary,
http://dragon.klte.hu/~gundat.
Present address:bDepartment of Clinical Chemistry, School of Medicine, University of Debrecen, Debrecen, Hungary
The chemistry of 2-substituted cephalosporins has never been a very intensively
probed area of b-lactam antibiotics. The main reason is
that according to early investigations these compounds possess only moderate
antimicrobial activities. However, modified derivatives of the well-known
ß-lactam antibiotics (penicillins, cephalosporins, carbapenems etc) may possess
two different, but very important biological activities: certain compounds can
inhibit the b-lactamase enzyme (this bacterial enzyme is responsible for the
destruction of the antibiotic in the resistant bacteria), 1 while others act on a completely different field: they
are inhibitors of the human leucocyte elastase (HLE) enzyme. This latter enzyme
of serine protease type is a key component of the body's inflammatory defenses by
participitating in the proteolytic degradation of microorganisms and tissues
during wound healing. Failed regulation of this enzyme activity may lead to a
number of diseases including rheumatoid arthritis, pulmonary emphisema, cystic
fibrosis etc.2
There are several structural differences between real antibacterial cephalosporin
antibiotics and HLE or b-lactamase enzyme inhibitors
(for examples click here), among others the latter
may be substituted in position 2, and the sulphur can be in a higher oxidation
state.
During our investigations for the functionalization of the C-2 position of
cephalosporins, we probed into the chemistry of 2-halogenocephalosporins. The
available literature data are quite scattering, and the results are not always
compatible. This is not surprising, as according to our and others' experiments
the results are strongly dependent on the reaction conditions used and the
structure of the starting compounds. The products are not always stable and we
came across several rearrangements leading to different non-ß-lactam products.
Discussion
Halogenation of cephalosporins
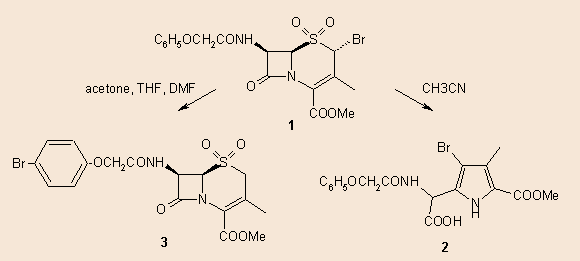
In our previous studies 3,4 we described the bromination of cephem sulphones and it was
found that 2-bromo-cephalosporin sulphones (1) easily rearrange to
bromopyrroles (2) or to the p-bromo-phenoxyacetyl derivative (3) on
standing in acetonitrile or other solvents:
In addition to spectroscopic evidences, the structure of the dimethyl ester of 2 was also proved by X-ray crystallography.
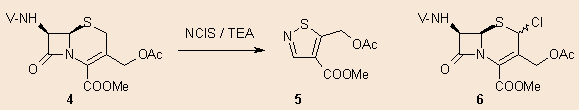
In this e-article we report our findings on the ionic chlorination of cephalosporins with NClS. In order to shed light on the influence of the oxidation state of the sulphur atom, we carried out the same reaction with the corresponding sulphides, sulphoxides and sulphones. Thus, if methyl 7-phenoxyacetamido-cephalosporanate (4) was allowed to react with N-chlorosuccinimide in CH2Cl2 in the presence of a catalytic amount of triethylamine, the only product we could isolate was the 5 isothiazoline derivative. This reaction was already described in the literature 7, stating that a mixture of the 2-chlorocephem and isothiazolone formed. In our case (with different compounds and reaction conditions) formation of the chloro derivative 6 could not be observed.
When the corresponding sulphoxide was treated with NClS under the same reaction conditions, no rearrangement occurred. Instead, the 2,2-dichloro derivative 8 formed. The monochloro derivative could not be prepared: even if NClS (1 eq.) was added very slowly to the reaction mixture, only 8 and unreacted starting material were observed:
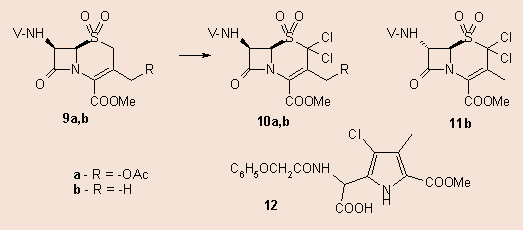
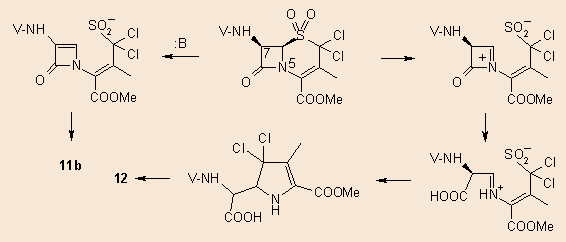
Similar reaction took place in the case of the 9a or b sulphone, only the 2,2-dihalogen derivatives 10a,b formed. A minor side-product (3-4%) was also found beside 10b in the reaction mixture, this was separated by column chromatography and proved to be the 7a-epimer of 10b. This base-catalyzed formation of 11b is characteristic of the sulphones of penicillins and cephalosporins. When the mother liquor after the separation of the first crop of crystals of 10b was evaporated, redissolved in acetonitrile and allowed to stay and evaporate slowly in the air, a new highly polar compound formed. According to spectroscopical evidences it 12, is the chlorine analogue of 2.
It is generally accepted, that the epimerization process giving rise to 11b proceeds via a b-elimination-addition of the sulphon moiety. This is a well-known base-catalyzed process among cephalosporin and penicillin sulphones and sulphoxides 5.
As to the formation of 12, we may speculate that the mechanism is similar to that suggested in our previous article.4 As shown in the above scheme, in this case 10b may also undergo a 1,5-bond cleavage, but now a positively charged immonium intermediate is produced. After hydrolytical opening of the ß-lactam ring the pyrrole formation takes place with simultaneous SO2 elimination. Subsequent HCl elimination leads to 12.
Reactions with mercury(II)trifluoroacetate
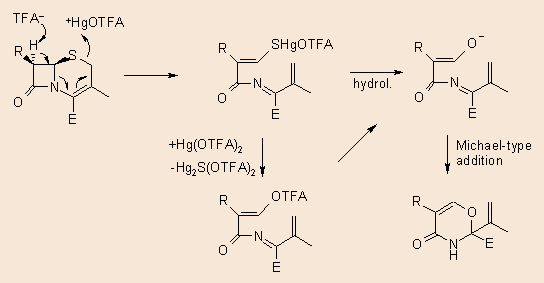
Several years ago we already examined the reaction of cephalosporins with mercury(II)trifluoroacetate and found, that the cephem esters 13 rearrange to the oxazolines 14: 6
Recently we are reinvestigating this reaction bearing in mind the possible mercuration at position 2, using the free cephem acid. When the 15 sulphide (n = 0) was reacted with Hg(II)trifluoroacetate in methanol, the main product isolated was a similar oxazolin, without the carboxylate function and containing a new methoxy substituent. The structure of the product as well as the fact, that 4 equivalents of the Hg-salt was necessary to complete the reaction, renders probable that a multistep, complex reaction is involved. The sulphoxide (15, n = 1) gave no reaction, while the sulphone (15, n = 2) yielded a complex reaction mixture, these investigations are in progress.
Our original assumption 6 for the formation of 14 is depicted in the following scheme. In the case of 16 there is certainly an additional oxydative decarboxylation, as well as an additional solvomercuration step. All suggestions for a possibly tentative mechanism are welcomed.
Experimental
Methyl 4-acetoxymethyl-isothiazol-3-carboxylate (5): 0.5 g (1.19 mmol) of 4 was dissolved in 10 ml of chloroform. 0.32 g (2.28 mmol) of N-chlorosuccinimide was added followed by 0.01 ml of triethylamine. According to tlc (tolueneˆEtOAc 1:1) all of the starting material has disappeared in 20 mins. The reaction mixture was diluted to 30 ml by adding chloroform, it was washed with 10% NaHCO3 solution and brine. After drying and evaporation the obtained row product was purified with chromatography on silica gel (tolueneˆEtOAc 3:1) yielding 0.19 g (75.6%) of 5, m.p. 90-93 ºC. - 1H NMR (200 MHz, DMSO-d6) d = 2.09 (s, 3 H), 3.87 (s, 3 H), 5.34 (s, 2 H), 9.09 (s, H). - Analysis C8H9NO4S (215.22) calcd. C 44.64, H 4.22, N 6.51, S 14.90; found C 44.6, H 4.2, N 6.5, S 14.9. - MS (EI) m/s 215 (M+), 172, 140, 43.
Methyl 3-acetoxymethyl-2,2-dichloro-7-phenoxyacetamido-ceph-3-em-4-carboxylate 1-oxide (8): 0.5 g of the sulphoxide 7 was allowed to react with 2 eqs. of NClS for 30 mins and worked up as described above. 0.32 g (55.1%) of 8 was obtained, m.p. 139-142 ºC. - 1H NMR (200 MHz, DMSO-d6) d = 2.05 (s 3 H), 3.90 (s, 3 H), 4.70 (s, 2 H), 4.74, 5.39 (ABq, H, J = 13.6 Hz), 5.69 (d H, J = 5.3), 6.21 (dd, H, J1 = 5.3, J2 = 8.5 Hz), 6.91 - 7.35 (m, 5 H), 8.57 (d, H, J = 8.6). - 13C NMR (DMSO-d6) d = 169.8, 168.4, 163.3, 160.3, 157.4, 129.5, 121.3, 114.5, 127.5, 116.6, 90.4, 66.1, 59.2, 66.5, 59.5, 53.7, 20.5.- IR (KBr) 1734, 1598, 1456, 1274, 1068 cm-1. - Analysis C19H18N2O8SCl2 (505.33) calcd. Cl 14.3, N 5.54, S 6.34; found Cl 14.2, S 6.5, N 5.6
Methyl 3-acetoxymethyl-2,2-dichloro-7-phenoxyacetamido-ceph-3-em-4-carboxylate 1,1-dioxide (10a): 1 g of the sulphone 9a was allowed to react with 2 eqs. of NClS for 30 mins and worked up as described above. 0.33 g (28.6%) was obtained after chromatographic purification, m.p. 115-117 ºC. - 1H NMR (200 MHz, DMSO-d6) d = 2.03 (s, 3 H), 3.86 (s 3 H), 4.62, 4.70 (ABq, 2 H, J = 15.3), 4.88, 5.11 (ABq, 2 H, J = 13.7), 6.22, (d, H, J = 5.1), 6.31 (dd, H, J1 = 5.1, J2 = 8.1), 6.85-7.31 (m, 5 H), 9.4 (d, H, J = 8.1). - Analysis C19H18N2O9SCl2 (521.33) calcd. Cl 13.6, N 5.37, S 6.15; found Cl 13.7, N 5.5, S 6.2.
Methyl 2,2-dichloro-3-methyl-7-phenoxyacetamido-ceph-3-em-4-carboxylate 1,1-dioxide (10b) and methyl 4-chloro-5-[carboxy-(2-phenoxyacetamido)methyl]-3-methyl-1H-pyrrole-2-carboxylate (12): 0.394 g (0.001 mol) of the sulphone 9b was allowed to react with 2 eqs. of NClS for 30 mins in CH2Cl2 and the row product was recrystallized from isopropanol giving rise to 0.17 g (36.7%) of first crop. After evaporation the residue was taken up in 5 ml of acetonitrile and was allowed to stay on room temperature. Tlc monitoring showed (tolueneˆEtOAc 1:1) that 9b has slowly disappeared and a new very polar compound appeared near the starting point. After 7 days the solvent was slowly allowed to evaporate to about 1 ml, the crystalls were collected and recrystallized from acetonitrile, yielding 10b: m.p. 135-136 ºC. - 1H NMR (200 MHz, DMSO-d6) d = 2.16 (s, 3 H), 3.80 (s, 3 H), 4.59, 4.75 (ABq, 2 H, J = 15.4 Hz), 6.19 (dd, H, J1 = 5.0 Hz, J2 = 8.2 Hz), 6.28, (d, H, J = 5.0 Hz), 6.86-7.32 (m, 5 H), 9.37 (d, H, J = 8.2 Hz). - Analysis C17H16N2O7SCl2 (463.29) calcd. Cl 15.3, N 6.05, S 6.92; found Cl 15.1, N 6.15, S 6.80. 12: m.p.: 217-222 °C (dec). - 1H NMR (200 MHz, DMSO-d6) d = 1.90 (s, 3 H), 3.81 (s, 3 H), 4.63 (s, 2 H), 6.89 (d, H, J = 4.8 Hz), 6.92-7.34 (m, 5 H), 7.81 (d, H, J = 4.8 Hz), 9.04 (s, H), 12.3 (broad s, H). - 13C NMR (200 MHz, DMSO-d6) d = 11.8, 55.2, 67.1, 104.8, 106.2, 115.2, 121.9, 130.0, 133.2, 133.2, 138.4, 138.7, 157.9, 164.7, 166.0, 167.5. - Analysis C17H17N2O6Cl (380.79) calcd. Cl 9.31, found Cl 9.43
2-Isopropenyl-6-methoxy-4-oxo-5-phenylacetamido-2H-[1,3]-oxazin (16): 3.6 g of red HgO was dissolved in 5 ml of trifluoroaceric acid with gentle warming. The clear solution was evaporated and dried azeotropically by the addition and evaporation of dry CH2Cl2. Finally it was dissolved in 10 ml of CH2Cl2. This solution was added rapidly to a solution of 1.8 g of 15 (n = 0) in 100 ml dry methanol. The mixture was refluxed for 6 hours, and after cooling H2S was bubbled through it for about half an hour. It was allowed to stay in the cool overnight. The precipitation was filtered, washed with methanol. The methanol solution was concentrated and chromatographed on silicagel (tolueneˆEtOAc 3:1). The main product was recrystallized from isopropanolˆether, yielding 0.38 g of 16. M.p.: 115-116 °C - 1H NMR (200 MHz, CDCl3) d = 2.02 (d, 3 H, J = 1.3 Hz), 5.56 (q, H, J = 1.3 Hz), 5.98 (s, H), 7.3-7.5 (m, 5H), 7.72 (br s, H), 7.83 (dd, H, J1 = 10.7 Hz, J2 = 0.7 Hz), 11.0 (d, H, J = 10.7 Hz). - 13C NMR (200 MHz, CDCl3) d = 18.3 (CH3), 44.3 (OCH2), 52.7 (OCH3), 108.5 (C), 122.2 (CH), 122.5 (CH2), 128.0 (CH), 129.3 (2 x CH arom.), 129.4 (2 x CH arom.), 133.6 (C), 138.7 (C), 165.1 (C arom.), 165.8 (CO), 170.1 (CO). - MS (EI) C16H18N2O4 (302.3306) m/s: 302 (M+), 271 (M+-OMe), 211, 184, 167, 140, 91, 69 (100%)
References
1. Reviews: a) O. A. Mascaretti, O. A. Roveri and G. O. Danelon: "Recent Advances in the Chemistry and Biochemistry of ß-Lactams as ß-Lactamase Inhibitors" in Recent Progress in the Synthesis of Antibiotics, (ed. Lukacs, G.), pp. 677-749, Springer Verl., Berlin, 1993; - b) R. Southgate, C. Branch, S. Coulton and E. Hunt: "Chemistry and Synthesis of Some Novel ß-Lactam Antibiotics and ß-lactamase Inhibitors" in Recent Progress in the Synthesis of Antibiotics, (ed. Lukacs, G.), pp. 621-675, Springer Verl., Berlin, 1993
2. Review: P. D. Edwards and P. R. Edwards, "Synthetic Inhibitors of Elastase" Med. Res. Rew. 1994, 14, 127
3. T. E. Gunda and G. N. Szöke: Synth. Comm.1997, 27, 3395
4. T. E. Gunda and G. N. Szöke: Tetrahedron, 1998, 54, 6565
5. See for example: a) C. M. Pant, R. J. Stoodley: J. C. S. Chem. Commun. 1978, 1366; b) P. J. Claes, G. DeCoster, L. A. Kerremans, H. Vanderhaeghe: J. Antibiotics, 1979, 32, 820
6. T. E. Gunda and C. Enebäck: Acta Chem. Scandinavica, 1980, B 34, 299
7. W. H. Koster, J. E. Dolfini and B. Toeplitz: J. Org. Chem. 1978, 43, 79