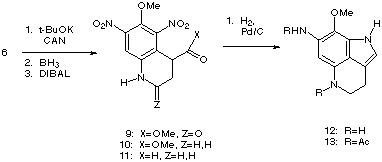Preparation of a Key Tricyclic Intermediate for the Synthesis of Pyrroloiminoquinone Natural Products
George A. Kraus* and Natesan Selvakumar
Department of Chemistry, Iowa State University, Ames, IA 50011
Fax 515-294-0105; e-mail: gakraus@iastate.edu
Summary: Indole 12 was prepared in seven steps from para-anisidine.
The key step was an intramolecular nucleophilic aromatic substitution reaction.
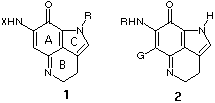
The group of natural products bearing a pyrroloiminoquinone subunit has
increased rapidly since the first compound was isolated in 1986.1
Among these compounds, the makaluvamines2 and the
batzellines3 contain the trisubstituted iminoquinone unit 1,
while veiutamine,4 the discorhabdins5 and
wakayin6 have the fully-substituted iminoquinone unit 2.
Veiutamine (2, R=H, G= p-hydroxybenzyl) group, exhibited activity
against solid tumors and was more active (IC50 0.3 mg/mL) than
makaluvamine D (1, R=H, X=p-hydroxyphenethyl) against human colon tumor
cell line HCT 116. The makaluvamines are topoisomerase II inhibitors.
Syntheses and several approaches have been reported.7 With the
exception of the route of Alvarez, Joule and coworkers (which begins with a
quinoline), most of the existing syntheses proceed by appending the B ring onto
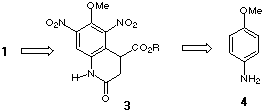
a bicyclic indole ring system. In our approach the C ring is appended onto a
tetrahydroquinoline ring system which in turn is made using a novel
intramolecular nucleophilic aromatic substitution reaction. Our synthesis
proceeds via intermediate 3 and is applicable to the preparation of both
1 and 2.
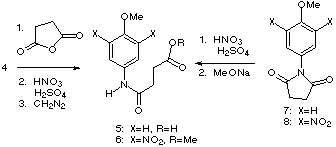
The initial synthesis of ester 6 began with the reaction of succinic
anhydride with para-anisidine (4). Nitration of the resulting amido
acid 5 using fuming nitric acid and sulfuric acid followed by
esterification provided 6 along with a variable amount of mono-nitration
product which made purification of 6 difficult. However, a modified
route beginning with succinimide 78 permitted a convenient
large scale synthesis of 6. Imide 7 underwent dinitration with
fuming nitric acid and sulfuric acid in 86% yield, affording 8 as a
yellow solid with mp 174 °C. The reaction of 8 with sodium
methoxide (1.2 equiv. in 6:1 THF:MeOH) provided amido ester 6 in 96%
yield.
With quantities of 6 now readily available, the intramolecular
nucleophilic addition reaction was investigated using various bases (LDA,
t-BuOK) in aprotic solvents.9 To the best of our knowledge,
intramolecular aromatic nucleophilic addition reactions similar to this one in
aprotic solvents have not been reported. Using three equivalents of potassium
tert-butoxide in THF at -35 °C followed by oxidative workup with one
equivalent of ceric ammonium nitrate (CAN) afforded lactam 9 in 55%
yield on a five millimole scale.10 Selective reduction of the
lactam using borane in THF from 0 °C to ambient temperature provided amine
10 in 84% yield. Partial reduction of the ester with DIBAL (13 equiv.
in toluene at -78 °C) afforded aldehyde 11 in 92%
yield.11 Reduction of the nitro groups using catalytic
hydrogenation in ethyl acetate over 12 hours provided indole 12 in 42%
yield.12 Catalytic hydrogenation in acetic anhydride with sodium
acetate afforded diacetate 13.
Tricyclic intermediate 12 can be prepared in only seven steps from
para-anisidine. The route is flexible and will make available quantities of
compounds for further biological evaluation. Interestingly, intermediate
9 should permit an additional nucleophilic addition, providing
convenient access to both makaluvamine and veiutamine analogs. References
(1) Molinski, T. F. Chem. Rev.1993, 93, 1825.
(2) Radisky, D. C.; Radisky, E. S.; Barrows, L. R.; Copp, B. R.; Kramer, R. A.;
Ireland, C. M. J. Am. Chem. Soc.1993, 115, 1632.
(3) Sun, H. H.; Sakemi, S.; Burres, N.; McCarthy, P. J. Org. Chem.1990, 55, 4964.
(4) Venables, D. A.; Barrows, L. R.; Lassota, P.; Ireland, C. M.
Tetrahedron Lett.1997, 38, 721.
(5) Perry, N. B.; Blunt, J. W.; Munro, M. H. G.; Higa, T.; Sakai, R. J.
Org. Chem.1988, 53, 4621.
(6) Copp, B. R.; Ireland, C. M.; Barrows, L. R. J. Org. Chem.1991, 56, 4596.
(7) White, J. D.; Yager, K. M.; Yakura, T. J. Am. Chem. Soc.1994, 116, 1831. Roberts, D.; Joule, J. A.; Bros, M. A.;
Alvarez, M. J. Org. Chem,1997, 62, 568. Yamada, F.;
Hamabuchi, S.; Shimizu, A.; Somei, M. Heterocycles,1995,
41, 1905. Peat, A. J.; Buchwald, S. L. J. Am. Chem. Soc.1996, 118, 1028. Sadanandan, E. V.; Pillai, S. K.;
Lakshmikantham, M. V.; Billimoria, A. D.; Culpepper, J. S.; Cava, M. P. J.
Org. Chem.1995, 60, 1800. Zhao, R.; Lown, W. Synth.
Comm.,1997, 27, 2103. Makoza, M.; Stalewski, J.;
Maslennikova, O. S. Synthesis,1997, 1131. Kita, Y.; Tohma, H.;
Inagaki, M.; Hatanaka, K.; Yakura, T. J. Am. Chem. Soc.1992,
114, 2175.
(8) Compound 7 was produced from 4 and succinic anhydride with
continuous removal of water. Piutti, A. Chem. Ber.1896,
29, 84.
(9) Nucleophilic aromatic substitution: Terrier, F. Chem. Rev.1982, 82, 77.
(10) A 2D COSY experiment on compound 10 confirmed the
tetrahydroquinoline structure.
(11) Fewer equivalents produced an inseparable mixture of starting material,
aldehyde and alcohol. The ratio of aldehyde to alcohol was 4:1.
(12) Compound 12: 300 MHz NMR (CDCl3) d 2.94 (t, J=5.7 Hz, 2
H), 3.43 (t, J=5.7 Hz, 2 H), 3.84 (s, 3 H), 5.79 (s, 1 H), 6.58 (s, 1 H), 7.66
(brs, 1 H). 75 MHz 13C NMR (CDCl3 +
CD3COCD3) d 23.0, 43.7, 59.8, 92.0, 111.2, 113.2, 113.5,
125.4, 128.1, 135.2, 137.6.
Compound 13: 300 MHz NMR (CDCl3) d 2.22 (s, 3 H), 2.46 (s,
3 H), 2.89-3.00 (m, 2 H), 3.98 (s, 3 H), 4.08-4.20 (m, 2 H), 6.84 (s, 1 H),
7.70 (bs, 1 H), 7.87 (bs, 1 H), 8.21 (bs, 1 H). MS m/z for
C15H17N3O3: 287, 230, 188. HRMS
m/z for C15H17N3O3: calcd.
287.1270; found 287.1267. Mp 217 °C with decomp.