| [Related articles/posters: 088 103 107 ] |
For the more detailed study of properties of KICl2 we presumed that it would be reasonable to use the specificy in structure and reactivity of phosphorylated allenes.
In addition, we tried to apply the synthetic performance of this reagent for development of methods of electrophilic iodocyclization of allenylphosphonates, which do not exist yet. The studied earlier reactions with I2, IBr and ICI required heating, and the yields of the final products were no higher than 50% [5]. Therefore the search of new iodinating agents seemed to be an actual synthetic task.
We found that dimethyl 3-methyl-1,2-butadienephosphonate (I) reacts with KIC12 in chloroform at room temperature in l5-20 min. After washing the reaction mixture with sodium thiosulfate we obtained 4-iodo-1,2-oxaphosphol-3-ene (II) in 80-85% yield. Its formation corresponds to the ordinary scheme of reactions of electrophilic reagents with such substrate, assuming that initial electrophilic attack occurs with iodine chloride. This fact well agrees with the common view on KICI2 as a source of ICl [1]. However, in the reaction with allene I is realised an earlier unknown type of reactivity of KICI2. As a minor product (yield about 10%), in the reaction mixture we detected dimethyl 3-methyl-2-chloro-2-butenephosphonate (III).
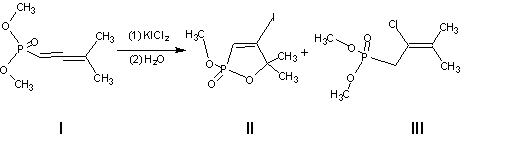
The regioselectivity of chlorine addition shows that compound III is formed as a result of nucleophilic attack of chloride anion at the a,b-double bond followed by protonation of the intermediate. Since the iodine chloride displays the properties of a Lewis acid [3], it seems possible that coordination at phosphoryl oxygen of the iodine chloride, formed intermediately from the original reagent, promotes addition of chloride anion. The alternative route with one-electron oxidation of the substrate with iodine chloride also may not be excluded.
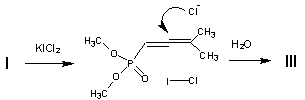
The possibility that the process proceeds by SET mechanism agrees with the recent report on probable reaction of ICl by this mechanism with aromatic substrates, also leading to chloro-substituted compounds [4].
In this step we did not attempted to elucidate unambiguously which mechanism acts predominantly in our case. Nethertheless, the fact that alternative reactivity of potassium dichloroiodate(I) actually exists illustrates spectacularly the possibilities to use the phosphorylated allenes as model substrates for the study of new reagents.
To obtain an additional information on both the reaction mechanism and its synthetic performance we studied reaction of KICl2, with other allenephos phonates, differing by substituents at the C1 atom of allene system and (or) at phosphorus atom. In this aspect, 1-alkoxy-2,3-pentadiene-2-phosphonates are particularly interesting due to their possible to allene- diene isomerization with formation of 3-halo-1,3- butadiene-2-phosphonates in the presence of hydrogen halides acids [5] or nucleophilic agents [6]. They are also capable to form cyclization products involv ing oxygen atom of alkoxymetylene fragment in the reactions with iodonium salts [7].
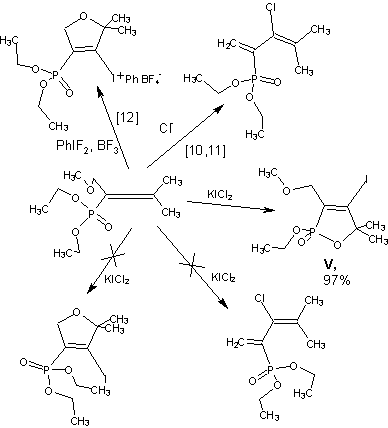
The reaction of diethyl 1-methoxy-2,3-penta-diene-2-phosphonates (IV) proceeds smoothly and leads to formation of a single product, 4-iodo-1,2-oxaphosphol-3-ene (V) only in almost quantitative yield.
It is noteworthy that neither allene-diene isomerization products nor the products of cyclization involving oxygen of methoxymethylene group are detected, unlike the reaction with iodonium salts [7], which obviously is caused by lower polarity of the reacting moiety.
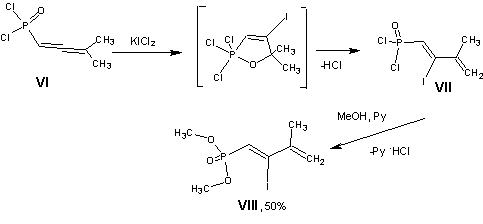
lodinating of 1,2-alkadienephosphonic dichlorides has not yet been described earlier. Therefore it seems that study of reaction of one of them with KICl2 is of particular interest.
Iodinating of 3-methyl-1,2-butadienephosphonic dichloride (Vl) with potassium dichloroiodate leads to 2-iodo-3-methyl- l,3-butadiehephosphonic dichloride (Vll) as the main product, what is typical for the re actions of allenephosphonic dichlorides with electro- philes [7].
Diene structure of compounds VII and VIll is confirmed by IR and NMR spectroscopy. In the 1H NMR spectrum of ester VIII there is doublet of the proton attached to C1 (d 6.4 ppm), and signals of terminal olefinic protons and the protons of methyl groups in an isopropenyl fragment. In the IR spectra occur absorption bands of two different double bonds (n 1590, 1640 cm-1).
According to 1H NMR spectrum, the reaction mixture also contains several unstable minor products formed probably as a result of addition at the 1-2 double bond of the allene system and by further transformation of the 1,3-alkadiene VII.
Thus, we developed a new approach to the synthesis of 4-iodo-1,2-oxaphosphol-3-enes and derivatives of 2-iodo-1,3-butadienephosphonic acids on the ground of reaction of KICl2 with allenephosphonic acid derivatives.
The unique combination in the allenephosphonate molecule of two double bonds essentially different in reactivity allowed us to discover an unusual reaction pathway involving KICl2.
The 1H NMR spectra were registered on a Varian, T-60 and Bruker WM-250 instruments with internal reference HMDS. Chromatography was carried out using Silufol UV-254 plates. The IR spectra were taken on a Specord 75IR spectrophotometer.
Reaction of potassium dichloroiodate(l) with dimethyl 3-methyl-1,2-butadienephosphonate (I). (a) A mixture of phosphonate (I) (0.176 g. I mmol) and KICl2, (0.237 g, 1 mmol) in 3 ml of anhydrous methylene chloride was stirred for 15 min at room temperature. After washing the reaction mixture with a solution of potassium thiosulfate it was treated with chloroform, the organic layer was dried over sodium sulfate, and the solvent was removed in vacuo. A mixture of 0.22 g (81%) 4-iodo-5,5-dimethyl-2-oxo-1,2-oxaphosphol-3-ene (II) and 0.02 g (10%) dimethyl 3-metbyl-2-chloro-2-butene-1-phosphonate (III) formed. After crystallization the mixture from heptane O.18 g (67%.) of pure phospholene II, mp 101 oC (7).
(b) The reaction was performed like in (a), but with ethanol as a solvent. We obtained O.170 g (63%) of iodide II, identical with that obtained in (a). Phospholene II. IR spectrum (film, n, cm-1): 1550 (C=C), l260 (P=O). 1H NMR spectrum (s, ppm): 6.5 d (1H, HC=C, JHP 26 Hz), 3.7 d (6H, OMe JHP, 12 Hz), 1.5 s (3H, Me), 1.45 s (3H, Me). 31P NMR spectrum, sP 34.7ppm.
Phosphonate III. IR spectrum: 1558 (C=C), 1260 (P=O). 1H NMR spectrum (s, ppm) 3 7 d (6H, OMe, JHP 12 Hz), 2.95 d (2H, CH2, JHP 22 Hz), 1.85 d (3H, Me, JHP 4.5 Hz). 1.80 d (3H, Me, JHP 7.0 Hz). 31P NMR spectrum. sP 24.5 ppm
Reaction of potassium dichloroiodate(l) wilh diethyl 1-methoxy-2,3-pentadiene-2-phosphonale (IV). Phosphonate IV (0.234 g, 1 mmol) and KICI2, (0.237 g, 1 mmol) were stirred for 15 min at room temperature, treated with aqueous solution of sodium thiosulfate (3 ml), then with chloroform. The organic layer was dried over sodium sulfate, and the solvent was removed m vacuo. 0.321 g (97%) of 5,5-dimethyl-4-iodo-3-methoxy and methyl-2-oxo-1,2-oxa-phosphol-1-ene (V) was obtained, mp 77ĽC (sublimed at 110 oC/2 mm). 31P NMR spectrum (CHCI3): dP 30.10 ppm. IR spectrum (mineral oil, n, cm-1): 1260 (P=O). 1H NMR spectrum, (s, ppm): 4.2 d (2H, CH2OMe, JHP14 Hz), 4.2- 3.8 m (2H, OCH2CH3), 3.4 s (3H, OMe), 1.5 s (3H. Me), 1.45 s (3H, Me), 1.2 t (3H, CH2CH3, J 7.0 Hz). Found. % C 31.93; H 5.23 C9H16IO4P. Calculated, % C 31.23, H 4.66.
Reaction of potassium dichloroiodate(l) with 3-methyl-1,2-buladienephosphonate dichloride (VI). To a solution of acyl dichloride Vl (2.55 g, 13.78 mmol) in 10 ml of anhydrous ethylene chloride while stirring at room temperature was added portionwise in 5 min 3.27 g (13.78 mmol) of KICI2. Stirrng was continued for 30 min (strong evolution of hydrogen chloride was observed), and the eluent was removed in vacuo. 3-Methyl-2-iodo-1,3. buudiene-1-phosphonic dichloride (VII) [1H NMR spectrum, d, ppm: 6.5 d (1H, HC=C), JHP, 14.0 Hz), 5.3 br.s (1H HC=C), 5.15 br.s (1H, HC=C), 2.1 s (3H, Me); 31P NMR spectrum sP 22.9 ppm] was treated without isolation with 2.2 ml (27.56 mmol) of dried pyridine and 3 ml of anliydrous methanol. After 15 min the mature was dissolved in 10 ml of chloroform, washed with aqueous solution of sodium thiosulfate (10 ml) an then with water (2 X 10 ml), dried over sodium sulfate. The solvent was removed, and after distilling the residue 2.1 g (50%) of dimethyl 2-iodo-3-methyl-1,3-butadiene-1-phosphonate (VII) was obtained, bp 133 oC (3 mm). IR spectrum (mineral oil, n, cm-1): 1640, 1590 (C=C-C=C), 1260 (P=O). 1H NMR spectrum, s, ppm: 6.4 d (1H, HC=C, JHP 14.0 Hz), 5.22 d.d. (1H, HC=C, J1, J2less than 1 Hz); 5.0 d.q (1H HC=C, J1 1.4 Hz, J2 less than 1 Hz), 3.7 d (6H. OMe. JHP 12.0 Hz), 2.0 m (3H, Me). 31P spectrum, sP 14.0 ppm. Found, % C 27.87, H 4.59; P 10.31. C7H12IO3P. Calculated, %. C27.84; H 4.00; P 10.26.
This work was performed under financial support of Russian Foundation for Basic Research (grant 96-03-33250).
1. Zefirov, N.S., Sereda, G.A., Sosonuk, S.S.,
Zyk, N.Y,, and Likhomanova, T.l., Synthesis,
1995, no. 11, pp. 1359-1361.
2. Angelov, Kh.M., Enchev, D.D., and Kirilov, M.,
Zh. Obshch. Khim., l 983, vol. 53, no. 9,
pp. 1958-1960.
3. Andrews, L.J. and Keefer, R.M. J. Org. Chem.,
1987, vol. 52, no. 13, pp. 2690-2694.
4. Turner, P.E., O'Malley, R.F., Sardella, D.J.,
Barinelli, L.S., and Kaul, P.J., J. Org. Chem.,
1994, vol. 59, no. 24, pp. 7335-7340.
5. Brel', V.K., Chunin, E.D., Dogadina, A.D.,
Skvortsov, N.K., Ignat'ev, V.M., and lonin, B.l.
Zh. Ohshch. Khim., 1980, vol. 50, no. 4,
pp. 770-773.
6. Brel', V.K. and Ahramkin, E.V., Zh. Ohshch.
Khim., 1991, vol. 6l, no. 4, pp. 770-773.
7.
Zefirov, N.S., Koz'min, A.S., Kasumov, T., Polekhin, K.A.. Sorokin, V.D., Brel, V.K., Ahramkin. E.V., Struchkov, Yu.T., Zhdankin, V.V,, and Stang, P.J., J. Org. Chem., 1992, vol. 57, no. 8, pp. 2433-2437.