| [Related articles/posters: 112 077 119 ] |
N-substituted maleimides are shown to react at moderately
high temperature with two equivalents of 3,6-disubstituted-s-tetrazine
to form 4,5-diazaphthalimides in a one-pot reaction, where the
s-tetrazine acts as both cycloaddition reagent and as oxidant. Facile
ring-opening of the maleimide ring occurs with nucleophiles; both
reactions have been exploited to produce linked di-(2'-pyridyl)-pyridazine
(DPP) ligand and bis-ligand systems.
Background
It was recognised by earlier pioneers in the field of s-tetrazine
chemistry that cycloadditions involving these reagents were distinctly
different from regular dienes.[1-6]
Nenitzescu [3] and Sauer [4]
were able to put the early reports of Carboni and Lindsey [5]
onto a quantitative basis; typically they found that 3,6-dicarbomethoxy-s-tetrazine
reacted with the electron-rich dienophile a-morpholinostyrene
4.7 x 105 faster than the electron-poor dienophile
acrylonitrile. Indeed, classical electron-poor dienophiles like
DMAD, diethyl azodicarboxylate, tetracyanoethylene and even the
archetype dienophile maleic anhydride failed to react. Clearly
the s-tetrazines were members of the inverse electron-demand
Diels-Alder dienes.
When we conducted reactions of 3,6-di(2'-pyridyl)-s-tetrazine
(DPT) 2a [7,8] with ring-strained
dienophiles such as 1 we noted that these dienophiles reacted
rapidly in chloroform solution at room temperature,[9]
however, maleimides, DMAD, benzoquinones and maleic anhydride
did not react under these conditions. Accordingly, when we were
generating highly-reactive isobenzofurans 4, by the s-tetrazine
induced fragmentation of 1,4-epoxynaphthalenes 1, it was
common to include an electron-poor dienophile such as the N-substituted
maleimide 6 in the reaction mixture at the start of the
reaction (Scheme 1).[9,10] This
was based on the knowledge that 6 was not reactive towards
the inverse electron-demand DPT 2a but would react with
the normal electron-demand isobenzofuran 4 as it was generated.
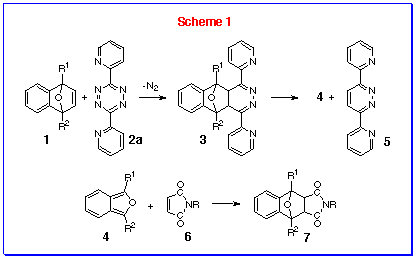
We had observed that the reaction of DPT 2a was slowed
down by introduction of substituents at the bridgehead position
of the 1,4-epoxynaphthalene 1, but a variety of substituents
could be tolerated, including electron-donating, electron-withdrawing
and sterically demanding groups like the trimethylsilyl group,
although the TMS derivative of 1 did require warming to
50 oC to ensure reaction. The corresponding 1-substituted
IBF 4 could be readily generated under these conditions
and trapped as their Diels-Alder adduct. N-methyl maleimide
6a was a common trapping agent as the singlet methyl resonance
was clearly detected in the 1H NMR spectrum and its
chemical shift characteristic of the exo v endo
stereochemistry of the products. When we sought a route to 1,3-bis(trimethylsilyl)
isobenzofuran by reacting 1,4-bis(trimethylsilyl)-1,4-epoxynaphthalene
with DPT 2a, we conducted the reaction in the presence
of N-methylmaleimide. Because no reaction occurred at room
temperature, the mixture was heated under reflux in toluene and
it was then that we discovered that N-methyl maleimide
6a reacted with DPT 2a.
Results and Discussion
We now report that this is a general reaction and that maleimides
can be made to react with s-tetrazine 2 at higher temperatures
to produce cycloadducts: further, s-tetrazine 2 takes
on a second role in this reaction, that of oxidant, thereby providing
access to N-substituted-4,5-diazaphthalimides 9 in
a one-pot reaction (Scheme 2).
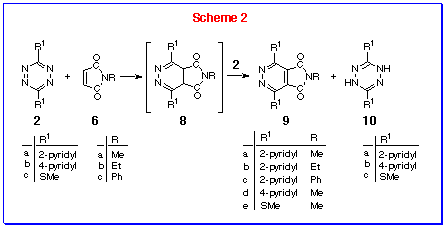
Reaction of DPT 2a with one equivalent of N-methyl
maleimide 6a in toluene at reflux produces a mixture of
products: the 4,5-diazaphthalimide 9a (50%), the dihydro-s-tetrazine
10a (50%) and unchanged maleimide 6a (50%). Increasing
the proportion of DPT 2a to two equivalents produces a
high yield of the 4,5-diazaphthalimide 9a and one equivalent
of reduced DPT 10a. This results is in accord with the
reaction pathway shown in Scheme 2, where the initially-formed
4,5-dihydropyridazine 8a (or its rearranged 1,4-dihydro-isomer)
is oxidised by DPT 2a to produce the observed 4,5-diazaphthalimide
9a.
The reaction is general and other maleimide (6b,c)
or s-tetrazine (2b,c) derivatives undergo
analogous reactions.
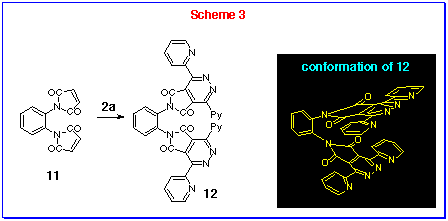
As the 4,5-diazaphthalimides 9a-c all contain the 3,6-di(2-pyridyl)-pyridazine (DPP) ligand, we also explored the potential of this reaction to produce linked bis-DPPs. Accordingly, the commercially available 1,2-bis(maleimido)-benzene 11 was reacted with DPT 2a under these vigorous conditions to form the linked bis-DPP 12, isolated in crystalline form m.p. 334-335 oC. The ortho-relationship of the DPP ligands brings the substituents too close to one another for them to be planar with the benzene ring and a twisted conformation must be adopted as shown in Scheme 3. The observation of total eleven carbons, including one carbonyl (d 163.65 ppm) and ten aromatic (d 124-155 ppm) carbons, could only be explained by this conformation due to its C2v symmetry.
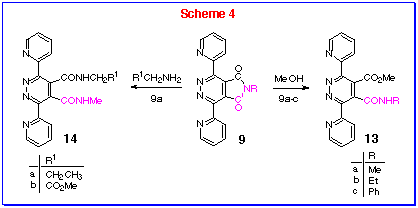
The maleimide ring in these 4,5-diazaphthalimides 9a-c
is especially prone to nucleophilic attack owing to the strong
electron-withdrawing effect of the pyridazine ring which is further
reinforced by the a-pyridyl substituents. Conversion to the ring-opened
product 13 occurs, for example simply by washing solid
9b with cold methanol in a filter funnel! Indeed, when
chromatographic separation of the phthalimides was conducted and
the eluant contained methanol (eg Method A, Experimental section),
the ring-opened products 13 (Scheme 4) were
obtained rather than the phthalimides 9. Similarly, n-propylamine
and glycine ethyl ester gave the ring-opened bis-amide 14a
and 14b, respectively. The reactions completed quickly
and were followed by the characteristic colour change, i.e.,
the bright-yellow reactants 9 turned to the colourless
products.
We also invested the reaction of DPT 2a with maleic anhydride
and found that no cycloaddition products were detected under the
above conditions. When prolonging reaction times or increasing
reaction temperatures were used we obtained charcolized reaction
mixtures from which dihydro-s-tetrazine 10a was
obtained as a major product. This suggested that DPT 2a
was being reduced, again indicative its role as an oxidising agent.
In order to assess its dehydrogenation ability DPT 2a was
reacted with acenaphthene in refluxing benzene, but no reaction
was observed (1H NMR monitor) after 46 hours; significantly
DDQ effects this dehydrogenation without difficulty. Aromatisation
of dihydro-s-tetrazine 10a using DDQ in deuteriochloroform
was very fast at room temperature and formation of DPT 2a
was complete within one minute (1H NMR monitor).
Experimental
Melting point were measured with a Gallenkamp melting pointing
apparatus in open capillaries and were uncorrected. Infrared spectra
were measured on a Perkin Elmer 1600 spectrometer using a pressed
potassium bromide disc. Mass spectra (EI, 70 ev) were recorded
on Shimadzu GC MS-QP2000A Spectrometer. Combustion microanalysis
were performed on Eager 200 instrument. High resolution mass spectra
(HRMS-EI) were recorded on a Micromass AutoSpec spectrometer at
70 ev. NMR spectra, 1H, 13C (Proton
Coupling and Proton Broad Band Decoupling), HMQC (1H-detected
Heteronuclear Multiple Quantum Coherence) experiments, were recorded
on a Bruker AMX 300 MHz Spectrometer in deuteriochloroform solution
(unless specified otherwise). The chemical shifts reported in
d units from internal reference, i.e. 0.000 ppm for tetramethylsilane
(TMS) in CDCl3, 2.050 ppm for CHD2COCD3
in acetone-d6, 3.350 ppm (1H) or 49.30 ppm
(13C) for CHD2OH in methanol-d4,
and 2.600 ppm (1H) or 39.50 ppm (13C)
for CHD2SOCD3 in DMSO-d6. The proton
assignment of 2'-pyridyl substituted groups was given in
Table 2 and 3 and omitted below. The starting compounds
were prepared by a standard method: 2a [7,11],
2c [12], 6c [13].
TLC was Merck TLC aluminium sheets silica gel 60 F254. Column
chromatography was operated on Merk silica gel 60.
Diels-Alder reactions of s-tetrazines and maleimides:
General Procedure: One equivalent N-substitutes
maleimide 6a,b,c with two equivalents of s-tetrazine
2a,b,c in sutable solvent was refluxed and stirred until
the typical violet colour of the s-tetrazine disappeared.
Bis-maleimide 11 required 4 equivalents of DPP 2a.
Products were obtained by removal of solvent under reduced pressure
followed by crystallisation or silica gel column chromatography.
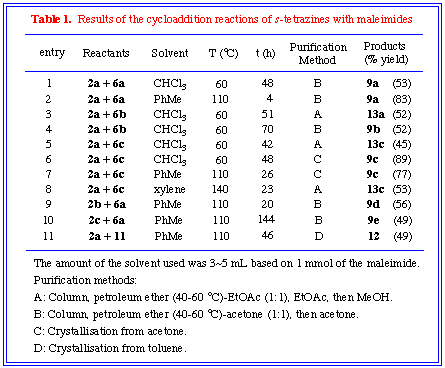
Reaction conditions, purification methods and results were summarised
in Table 1.
Nucleophilic addition on 4,5-diazaphthalimides 9a-c:
General Procedure: 4,5-diazaphthalimides 9a-c was
stirred at room temperature for one to two hours with excess nucleophile,
i.e. methanol, n-propylamine or glycine ethyl ester
(in dichloromethane solution). Concentration and crystallisation
from methanol (for 13a-c) or acetone (for 14a,b)
provided crystalline products in high yield (95-100%).
9a yellow needle from acetone, m.p. 222.4-223.8 oC.
IR (cm-1): 1724 (s, CO), 1586 (m), 1436 (m), 1374 (m),
1362 (m), 1270 (m), 1014 (m), 992 (m), 798(m), 752 (m); 1H
NMR: 3.217 (s, 3H, NCH3); 13C NMR: 24.85
(NCH3), 124.95 (Py-C3'/3''), 125.09
(Py-C5'/C5''), 127.78 (C4/5), 136.85 (Py-C4'/C4''),
149.71 (Py-C6'/C6''), 152.06 (Py-C2'/C2''),
155.27 (C3/6), 165.48 (CO); EIMS m/e (relative intensity)
317 (97), 289 (25), 260 (10), 246 (20), 218 (21), 205 (35), 206
(45), 204 (100), 203 (49), 178 (11), 177 (21), 176 (24), 152 (15),
151 (23), 150 (14), 138 (10), 102 (20), 78 (47). Anal. Calcd for
C17H11N5O2.0.25H2O:
C, 63.45; H, 3.60; N, 21.76. Found: C, 63.04; H, 3.31; N, 21.76.
9b yellow needle from acetone, m.p. 197.9-198.9 oC.
IR (cm-1): 1734 (s, CO-sym.), 1717 (s, CO-asym.), 1587
(m), 1441 (m), 1395 (m), 1375 (m), 1347 (m), 1047 (m), 993 (m),
800 (m), 754 (m); 1H NMR: 1.282 (t, 3H, J=7.23
Hz, NCH2CH3), 3.782 (q, 2H, J=7.23
Hz, NCH2CH3); (acetone-d6):
1.231 (t, 3H, J=7.20 Hz, NCH2CH3),
3.717 (q, 2H, J=7.20 Hz, NCH2CH3);
13C NMR: 14.14 (NCH2CH3),
34.79 (NCH2CH3), 125.70 (Py-C3'/3''),
125.73 (Py-C5'/5''), 128.40 (C4/5), 137.54 (Py-C4'/4''),
150.31 (Py-C6'/6''), 152.69 (Py-C2'/2''),
155.90 (C3/6), 165.94 (CO); EIMS m/e (relative intensity)
331 (100), 303 (20), 274 (11), 246 (23), 204 (70), 176 (11), 151
(11), 102 (13), 78 (30). Anal. Calcd for C18H13N5O2:
C, 65.25; H, 3.95; N, 21.14. Found: C, 65.08; H, 3.87; N, 21.16.
9c yellow needle from acetone, m.p. 304-306 oC (fast heating).
IR (cm-1): 1734 (s, CO), 1566 (w), 1492 (m), 1384 (m),
1374 (m), 1129 (m), 991 (w), 799 (m), 759 (m); 1H NMR:
7.361-7.532 (m, 5H, 5xPhCH); 13C NMR: 125.76 (Py-C3'/3''),
125.81 (Py-C5'/5''), 127.45 (2xPh-ortho),
128.01 (C4/5), 129.48(Ph-para), 129.85 (2xPh-meta),
131.64 (Ph-ipso), 137.63 (Py-C4'/4''), 150.31
(Py-C6'/6''), 152.52 (Py-C2'/2''),
156.18 (C3/6), 165.09 (CO); EIMS m/e (relative intensity)
379 (100), 322 (10), 204 (70), 144 (15), 78 (18). Anal. Calcd
for C22H13N5O2: C,
69.64; H, 3.45; N, 18.46. Found: C, 69.87; H, 3.30; N, 18.50.
9d yellow crystal from acetone, m.p. 205.9-206.7 oC.
1H NMR: 3.29 (s, 3 H, NCH3), 8.05 (d, 4
H, J=3.6 Hz, Py-H3'/5' and Py-H3''/5''),
8.91 (d, 4 H, J=3.6 Hz, Py-H2'/6' and Py-H2''/6'');
13C NMR: 25.79, 124.85, 127.05, 140.62, 150.91, 154.57,
166.52; HRMS-EI calcd for C17H11N5O2:
317.0913, found 317.0912.
9e orange crystal from acetone, m.p. 292.4-293.9 oC.
1H NMR: 2.83 (s, 6 H, 2xSCH3), 3.19 (s,
3 H, NCH3); 13C NMR: 13.14 (SCH3),
24.82 (NCH3), 125.23 (C4/5), 155.05 (C3/6), 167.12
(CO); HRMS-EI calcd for C9H9N3O2S2:
255.0136, found 255.0137.
12 yellow powder from acetone, m.p. 333.5-334.5 oC (decomp.).
IR (cm-1): 1748 (s, CO), 1588 (w), 1506 (m), 1391(m),
1380 (m), 998 (w); 1H NMR: 7.573-7.592 (m, 4H, 4xPhCH);
(DMSO-d6): 7.677-7.739 (m, 6H, 2xPhCH and 4xPy-H5), 7.764-7.820
(m, 2H, 2xPhCH); 13C NMR (DMSO-d6): 124.90,
124.94 (Py-C5/3), 127.59, 128.65 (Ph-ortho and Ph-meta),
129.65, 129.89 (Ph-ipso and pyridazine-C4/5), 136.71 (Py-C4),
149.44 (Py-C6), 151.51 (Py-C2), 154.64 (pyridazine-C3/6), 163.65
(CO). Anal. Calcd for C38H20N10O4.2H2O:
C, 63.69; H, 3.38; N, 19.54. Found: C, 63.69; H, 3.23; N, 19.07.
13a colourless rod from methanol, 100% yield. m.p. 191 oC (decomp., fast heating).
IR (cm-1): 3312 (sharp, m, NH), 1736 (s, CO-O), 1674
(s, CO-N), 1584 (m), 1570 (m), 1560 (m), 1378 (m), 1271 (m), 1203
(m), 1078 (m), 996 (m), 974 (m), 797 (m), 746 (m); 1H
NMR: 2.950 (d, 3H, J=4.86Hz, NCH3), 3.889 (s,
3H, OCH3), 6.485 (br q, 1H, J=4.86Hz, NH); 13C
NMR: 26.82 (NCH3), 52.94 (OCH3), 123.19,
124.15, 124.61, 124.87 (Py-C5'/5'', Py-C3'/3''),
130.01, 133.02 (C4/5), 137.11, 137.23 (Py-C4'/4''),
148.73, 148.93 (Py-C6'/6''), 152.59, 153.69 (Py-C2'/2''),
154.47, 155.52 (C3/6), 165.30, 166.51 (2xCO); EIMS m/e
(relative intensity) 349 (8), 348 (18), 320 (38), 319 (83), 318
(27), 317 (51), 291(18), 288 (19), 277 (16), 206 (43), 205 (89),
204 (55), 203 (26), 178 (11), 177 (11), 176 (12), 152 (13), 151
(15), 144 (11), 130 (15), 103 (28), 102 (24), 78 (100). Anal.
Calcd for C18H15N5O3:
C, 61.89; H, 4.33; N, 20.05. Found: C, 61.80; H, 4.25; N, 20.07.
13b colourless rod from methanol, 100% yield. m.p. 177.6-178.6 oC (decomp.).
IR (cm-1): 3244 (sharp, s, NH), 1733 (s, CO-O), 1668
(s, CO-N), 1589 (s), 1576 (m), 1546 (m), 1429 (s), 1384 (s), 1304
(s), 1286 (s), 1275 (s), 1200 (s), 996 (m), 963 (m), 780 (m);
1H NMR: 1.217 (t, 3H, J=7.26Hz, NCH2CH3),
3.703 (dq, 2H, J=7.26Hz, 5.88Hz, NCH2CH3),
3.906 (s, 3H, OCH3), 6.408 (br t,1H, J=5.88Hz,
NH); (acetone-d6): 1.209 (t, 3H, J=7.26Hz, NCH2CH3),
3.376-3.467 (m, 2H, NCH2CH3), 3.918
(s, 3H, OCH3), 7.505 (br s,1H, NH); 13C
NMR: 15.02 (NCH2CH3), 35.69 (NCH2CH3),
53.51 (OCH3), 123.82, 124.79, 125.25, 125.52 (Py-C5'/5'',
Py-C3'/3''), 130.62, 133.71 (C4/5), 137.76, 137.88
(Py-C4'/4''), 149.37 (two peaks, distance <
0.01 ppm, Py-C6'/6''), 153.24, 154.40 (Py-C2'/2''),
155.10, 156.22 (C3/6), 164.98, 167.09 (2xCO); EIMS m/e
(relative intensity) 363 (7), 331 (100), 319(97), 303 (22), 291
(16), 288 (39), 246 (22), 204 (70), 177(12), 176 (13), 152 (16),
144 (31), 102 (27), 78 (78). Anal. Calcd for C19H17N5O3:
C, 62.80; H, 4.72; N, 19.27. Found: C, 62.76; H, 4.69; N, 19.30.
13c colourless rod from methanol, 100% yield. m.p. 204 oC (decomp., fast heating).
IR (cm-1): 3246 (m, NH), 1735 (s, CO-O), 1661 (s, CO-N),
1608 (s), 1546 (m), 1448 (m), 1408 (m), 1382 (m), 1322 (m), 995
(m), 793 (m), 746 (m); 1H NMR: 3.871 (s, 3H, OCH3),
7.173 (tt, 1H, J=7.41, 1.14Hz, PhCH-para), 7.339-7.430
(m, 4H, 2xPhCH-meta and 2xPy-H5), 7.540-7.573 (dm, 2H,
J=7.53Hz, 2xPhCH-ortho), 8.513(br s, 1H, NH); (acetone-d6):
3.781 (s, 3H, OCH3), 7.119 (tt, 1H, J=7.38,
1.17Hz, PhCH-para), 7.305-7.371 (tm, 2H, J=7.95Hz,
2xPhCH-meta), 7.585-7.614 (dm, 2H, J=6.87Hz, 2xPhCH-ortho),
9.451(br s, 1H, NH); (methanol-d4, ppm): 3.389 (s, 3H,
OCH3), 7.142 (tt, 1H, J=7.41, 1.20Hz, PhCH-para),
7.288-7.354 (m, 2H, 2xPhCH-meta), 7.526-7.572 (m, 3H, 2xPhCH-ortho
and 1xPy-H5); 13C NMR (methanol-d4): 122.31
(Ph-ortho), 125.20, 125.56, 126.00, 126.15, 126.16 (Ph-para,
Py-C3'/3'' and Py-C5'/5''), 130.02
(Ph-meta), 132.64 (Ph-ipso), 138.84, 138.92(Py-C4'/4''),
139.25, 139.57 (C4/5), 150.43, 150.45 (Py-C6'/6''),
155.27, 156.03 (Py-C2'/2''), 156.94, 157.28 (C3/6),
171.21, 167.16 (2xCO); EIMS m/e (relative intensity) 411
(<0.1) 379 (77), 319 (100), 291 (21), 204 (59), 177 (7), 152
(5), 144 (19), 102 (6), 78 (35). Anal. Calcd for C23H17N5O3.CH3OH:
C, 65.00; H, 4.77; N, 15.79. Found: C, 64.82; H, 4.71; N, 15.84.
14a colourless crystal from acetone, 100% yield, m.p. 209.4-211.0 oC.
1H NMR: 0.94 (t, 3 H, J=7.3 Hz), 1.53-1.62 (m,
2 H), 2.91 (d, 3 H, J=4.9 Hz), 3.30 (q, 2 H, J=6.7
Hz), 6.87 (t, 1 H, J=5.3 Hz), 7.02 (q, 1 H, J=4.5
Hz), 7.27-7.31 (m, 2 H), 7.76-7.81 (m, 2 H), 8.23 (d, 2 H, J=8.0
Hz), 8.47 (d, 1 H, J=4.5 Hz), 8.50 (d, 1 H, J=4.4
Hz). 13C NMR: 12.12, 22.96, 27.39, 42.60, 124.47 (2xC),
125.09 (2xC), 133.24, 133.31, 137.58 (2xC), 149.20, 149.40, 154.08,
154.15, 155.58 (2xC), 165.85, 166.58; HRMS-EI calcd for C20H20N6O2:
376.1648, found 376.1651.
14b colourless crystal from acetone, 95% yield, m.p. 197.5-198.9 oC.
1H NMR: 1.25 (t, 3 H, J=7.1 Hz), 2.85 (d, 3
H, J=4.7 Hz), 4.09 (d, 2 H, J=5.3 Hz), 4.15 (q,
2 H, J=7.1 Hz), 7.18 (br m, 2 H), 7.46 (q, 1 H, J=4.7
Hz), 7.64-7.71 (m, 2 H), 7.75 (t, 1 H, J=5.3 Hz), 8.06-8.10
(m, 2 H), 8.32 (d, 1 H, J=4.4 Hz), 8.40 (d, 1 H, J=4.4
Hz); 13C NMR: 14.78, 27.44, 42.47, 61.87, 124.15, 124.62,
124.88 (2xC), 132.43, 133.44, 137.43, 137.51, 149.07, 149.31,
153.57, 153.99, 154.87, 155.34, 165.98, 166.21, 169.59; HRMS-EI
calcd for C21H20N6O4:
420.1546, found 420.1554.
Spectral Analysis
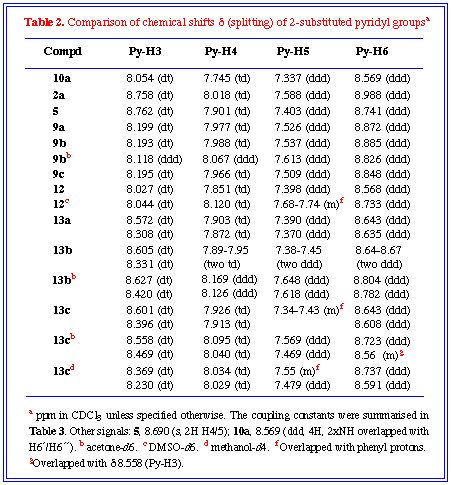
1H NMR: The typical splitting signals for the 2'-pyridyl
groups were observed at low field which made the total assignment
very easy. The 4,5-diazaphthalimides 9a-c showed almost
identical 1H NMR spectra due to the same dipyridyl
pyridazine moiety. The methanol addition products 13a-c
showed two sets of pyridyl resonances in the aromatic region due
to the two different environments of the 2'-pyridyl groups. The
order of chemical shifts were in agreement with 2-substituted
pyridines a>g>b,[14] i.e.,
py-H6 > py-H4 > py-H5, with the only exception of py-H3.
(Table 2 and Table 3). The remarkable increase of
chemical shift at pyridine H3 is ascribed to the deshielding effect
of pyridazine or (s-tetrazine ring for 2a and 10a).
The total assignment of protons and carbons were achieved by 1H,
13C (broad band decoupling and coupling) and HMQC NMR
techniques.
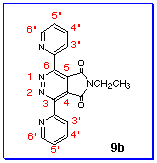
The difficult assignment of carbon signals was three quaternary
carbons (C-2'/2'', C-3/6, C-4/5). With compound
9b as the example, the signals were d 155.90 ppm, d 152.69
ppm and d 128.40 ppm. From the structure, d C-3/6 should be similar
to and slightly larger than d C-2'/2'' because
pyridazine ring was more electron-deficient ring than pyridine.
Furthermore, d C-3/6 should be larger than d C-4/5 because of
the inductive effect of nitrogen atom and conjugated effect of
carbonyl group in maleimide moiety.[15]
So the predicted order of chemical shifts was d C-3/6 > d C-2'/2''
> d C-4/5, i.e., C-3/6 155.90 ppm, C-2'/2''
152.69 ppm and C-4/5 128.40 ppm. The assignment proved to be correct
by proton coupling 13C NMR spectrum, in which 152.69
ppm appeared as double doublet peak (11.55 Hz and 7.08 Hz) because
C-2'/2'' coupled with H-4'/4'',
H-6'/6'' by 3J, 155.90 ppm was
a broad single peak (W0.5=4 Hz) because C-3/6 had a
long-range coupling (3J) with H-3'/3''
and 128.40 ppm was a sharp peak because C-4/5 had no coupling
protons.
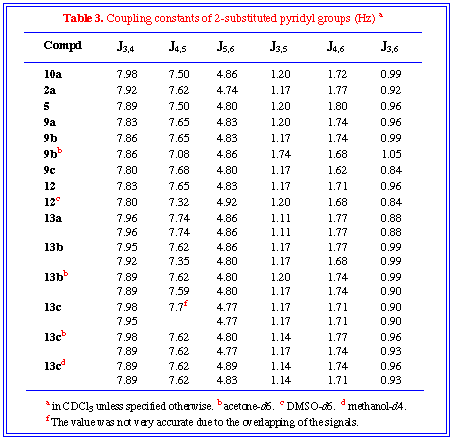
The magnitude of interproton coupling constants was consistent
with that for heterocyclic compounds, in which nitrogen atom lead
to a decrease in Jortho across the adjacent bond, i.e.,
J5,6 (ca. 4.8 Hz) was smaller than J3,4
and J4,5 (7-8 Hz).[16]
(see Table 3)
In the IR spectra, 4,5-diazaphthalimides 9 and 12
showed one strong carbonyl absorption at 1730-1750 cm-1.
The higher frequency of amide carbonyl was due to ring strain
from five member ring and electron-withdrawing effects of the
fused heterocyclic ring. At this region, ring opening products
(13) showed two bands 1730-1750 and 1660-1690 cm-1,
assigned to ester and amide carbonyls respectively. Other typical
absorption was C-H bending of methoxy at 1383 cm-1
(strong) and N-H stretching at 3244 cm-1 (sharp). All
the compounds showed typical absorption near 1000 cm-1
due to 2-monosubstituted pyridine ring breathing.[17]
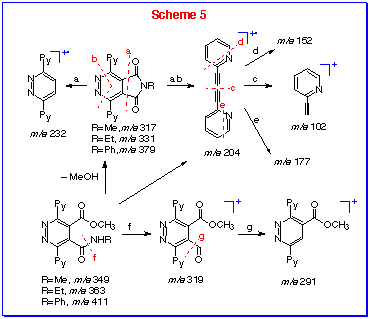
The mass spectra showed the cycloaddition products (9)
were very stable, molecular peak was base peak (9b,c)
or second strongest peak (9a). The methanol addition products
13 showed typical fragmentation ions at m/e 319
(M-NHR) and m/e 291 (M-NHR-CO). This indicates that the
C-N bond of the amide is weaker than C-O bond of ester. Characteristically,
the fission via thermal elimination of methanol gave the
cyclic 9 as a strong fragment. Both structures 9
and 13 favoured the loss of nitrogen and maleimide moiety
to form the fragment m/e 204 (path ab) (Scheme 5).
Acknowledgment
The authors thank Mrs Sitalakshmy Sankar for preparing 1,3-bis(trimethylsilyl)-1,4-epoxynaphthalene
and treating it with DPT 2a in the presence of N-methyl
maleimide which led to the original observation, the Central Queensland
University for financial support and the Australian Research Council
for the award of a Senior Research Fellowship to R.N.W. (1992-1996)
References
(1) For comprehensive review see: Boger, D. L. and Weinreb, S. N.; In Hetero Diels-Alder Methodology in Organic Synthesis, Academic Press, Inc., San Diego, 1987; p335-348 and references therein.
(2) Gilchrist, T.L.; In Heterocyclic Chemistry, Longmann
Scientific and Technical, New York, 2nd ed., 1992; p272-273.
(3) Avram, M, Dinulescu, J. G., Marica, E., Nenitzescu, C. D.
Chem. Ber. 1962, 95, 2248.
(4) For a recent review see: Sauer, J. Bull. Soc. Chim. Belg.,
1992, 101, 521.
(5) Carboni, R.A.; Lindsey, R.V. Jr. J. Am. Chem. Soc.,
1959, 81, 4342.
(6) Boger, D. L.; Sakya, S. M. J. Org. Chem., 1988,
53, 1415.
(7) Geldard, J. F.; Lions, F. J. Org. Chem., 1965,
30, 318.
(8) One of the earliest use of 3,6-di(2-pyridyl)-s-tetrazine
2a acting as an inverse electron-demand diene was reported
by Wilson, W. S.; Warrener, R. N. Tetrahedron Lett., 1970,
4787.
(9) Warrener, R. N. J. Am. Chem. Soc., 1971, 93,
2346.
(10) Warrener, R. N.; Evans, D. A. C.; Paddon-Row, M. N., Russell,
R. A. Aust. J. Chem., 1982, 34, 757.
(11) Russell, R.A.; Longmore, R.W.; Warrener, R.N. J. Chem.
Ed., 1992, 69, 164.
(12) Sandstrom, J. Acta Chem.Scand., 1961,
15, 1575.
(13) Fieser, L.F.; Fieser, M. In Reagents for Organic Synthesis,
John Wiley and Sons, Inc., New York, Vol. 1, 1967; p 845-846.
(14) Kemp, W. In Organic Spectroscopy, 3rd ed., MacMillan
Education Ltd., Houndmills, 1991; p 175.
(15) Wehrli, F.W.; Marchand, A.P.; Wehrli, S. Interpretation
of Carbon-13 NMR Spectra, 2nd ed., 1988, John Wiley
and Sons Ltd., Chichester, p 57-63.
(16) Jackman, L.M.; Sternhell, S. Applications of Nuclear Magnetic
Resonance Spectroscopy in Organic Chemistry, 2nd ed., 1969,
Pergamon Press, Oxford, p 305-310.
(17) Katritzky, A.R. In Physical Methods in Heterocyclic Chemistry,
Academic Press, New York, Vol. 2, 1963; p 274-303.