| [Related articles/posters: 107 103 088 ] |
Introduction, Results and Discussion, Experimental, References
Introduction
The BLOCK coupling methodology [1]
recently developed by our group uses preformed alkene building
BLOCKS with particular inbuilt effector groups (eg, ligands, chromophores,
binding agents, intercalators)[2] and
joins them together to form large rigid structures. Such BLOCKS
are usually polyalicyclic molecules with precisely defined geometries
and able to take part readily in cycloaddition reactions. These
properties make BLOCKS ideal building units when the design of
products of exactly defined size and shape is required.
The coupling process is usually stereoselective and often
produces only one product. The observed exo-stereoselectivity
is not surprising because almost all of these coupling methods
utiltise norbornene-fused BLOCKS as one of the cycloaddends and
from theoretical and experimental reports, it is known that norbornene
shows exo-stereoselectivity in most reactions, particulary
cycloadditions.[3]
The norbornene coupling methods can be divided into two major
groups depending on whether the two BLOCKS
react directly, or another compound
is necessary to act as a "glue" between the two BLOCKS.
Special attention will be paid to the newly developed methodology
for photochemical dimerisation of BLOCKS containing a cyclobutene diester which is described
in this paper for the first time.
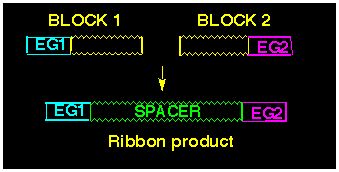
Direct Joining of Two BLOCKS.
The methodology used for the direct joining of two BLOCKS bearing
effector groups EG1 and EG2 is shown schematically
in Diagram 1. The polyalicyclic sections of the two BLOCKS become
connected together to produce the "spacer" part of the
ribbon-like product, thereby placing the effector groups at each
end of the molecule.
Two methods have already been developed in our laboratories which
can be used to produce this type of coupled product (viz ACE and aza-ACE
coupling), and a third one (photodimerisation) is the subject
of the present paper:
Photodimerisation of Cyclobutene Diester
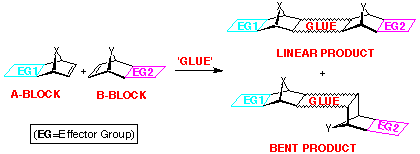
Joining BLOCKs by using various "GLUES"
The second approach involves the use of so-called "glues",
eg compounds which react with norbornene-fused BLOCKs in such
a way that part of the glue remains inbuilt in the product (Scheme
1). Extended-frame products usually result from these
coupling methods, although bent-frame geometries have
been observed in some cases when oxadiazole coupling is employed.
In this paper we outline three "glue" coupling methods:
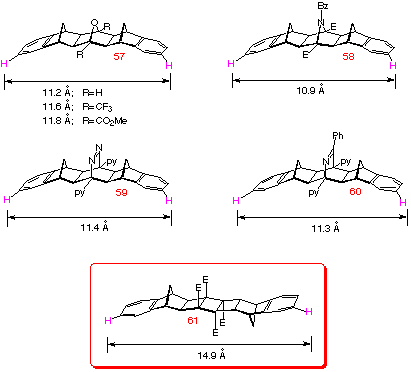
1,2,4-Triazine Coupling Method.
Click here to view geometrical
comparison of AM1 structures of products obtained by different
coupling procedures).
Photodimerisation of 1,2-Cyclobutene
Diesters[4]
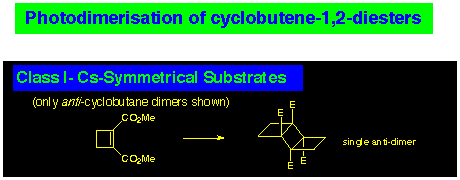
The photodimerisation of cyclobutene 1,2-diesters can be classified into three classes, depending on the nature of the frame to which they are attached. The simplest of these is where the substrate has Cs-symmetry (CLASS I) (Scheme 1). In this case, only two cyclobutane products are possible: a syn-dimer and an anti-dimer, with the latter the most commonly observed product.
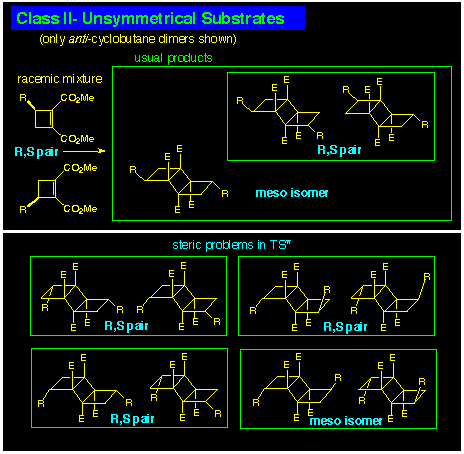
The CLASS II reaction is the most complicated as the 3-substituted
cyclobutene-1,2-diester substrates used to illustrate the outcome
of dimerisations in this class is a racemate and dimerisation
can mix and match between enantiomers. Theoretically a total
of 20 photodimers are possible; 10 anti-dimers and 10 syn-dimers.
The set of anti-dimers is shown in Scheme 2. Fortunately
7 of the possible 10 are excluded from practical consideration
owing to bad steric interactions in the transition state for dimerisation
(Diagram 3). Notwithstanding, the best result is formation
of a pair of enantiomeric dimers together with the meso isomer where it
is assumed that only anti-dimerisation occurs and the steric TS
restrictions are valid.
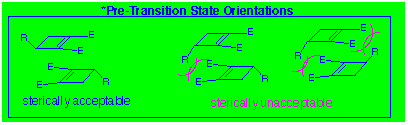
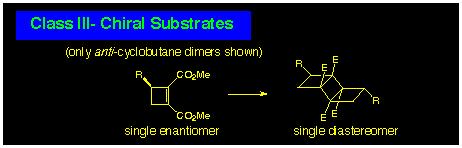
The CLASS III reaction involves dimerisation of a single enantiomer.
This is illustrated by the 3-substituted cyclobutene-1,2-diester
system where only two cyclobutane photoproducts, a single anti-dimer
and a single syn-dimer, need be considered. In practice,
anti-cyclobutane formation is usually observed and so a
single product of fixed stereochemistry is produced in this class
of photodimerisation.
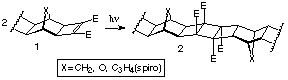
We have used the ability of dimethyl cyclobutene-1,2-dicarboxylates
to undergo [2p+2p]
self-dimerisation under photochemical conditions to build large,
ribbon-like structures (Scheme 4) and the examples reported herein
encompass all three CLASSES of reaction types.
Preparation of the cyclobutene diester building BLOCKS for this type of assembly is
well documented and one very useful method we have used
extensively in our work utilises Mitsudo's ruthenium-catalysed
[2p+2p]
cycloaddition of dimethyl acetylenedicarboxylate (DMAD) to the
double bond of norbornenes.[5] Other
methods include the related Lewis acid-catalysed cycloaddition
of DMAD or propiolates to alkenes [6]
and bishomo Diels-Alder addition of DMAD and related electron-deficient
acetylenes to quadricyclanes[11].
Literature Examples of Cyclobutene-1,2-diester Photodimerisation
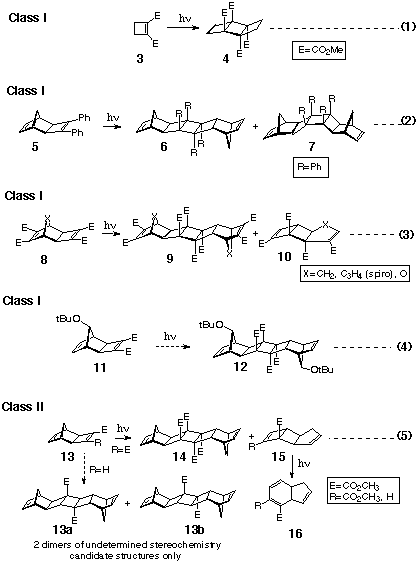
The first dimerisation of this type was reported in 1964 when
Seebach revealed that ultraviolet irradiation of solutions of
dimethyl cyclobutene-1,2-dicarboxylate (3) formed a dimeric
product assigned the anti-stereostructure 4 [7],
see equation 1 (Scheme 5). Eberbach has shown that the photodimerisation
reaction could be applied to polyalicyclic-fused cyclobutene-1,2-dicarboxylates
(equation 5, Scheme 5) and to 1,2diphenyl-cyclobutenes (equation
3, Scheme 5),[4a] where special attention
has been directed to the stereochemistry of the photodimerisation
products. Thus, Smith's diene 13 (R=CO2Me)
afforded a single anti-dimer 14 (R=CO2Me)
(CLASS I reaction), whilst the related monosubstituted methyl
cyclobutene-1-carboxylate 5 (R=H) (CLASS II reaction) formed
in low yield (<10%) a mixture of isomeric dimers of undetermined
structure see equation 5 (perhaps 13a and 13b) (Scheme
5). Both reactions produced an isomerised by-product 15,
which upon further irradiation was transformed into the corresponding
cyclohexadiene derivative 16.
The tricyclic compound 8 which contains two different types
of ester-substituted double bonds, produced analogues cyclobutane
dimers originating from site selective reaction at the cyclobutene
p-centre, see equation 3 (Scheme 5).
This coupling method has also been used by our own group to produce
spacer molecules based on the dimer 12 from irradiation
of 7-tert-butoxy Smith's diene 11 - equation 4 (Scheme
5).[4b, 4c]
The Synthesis of Novel Photodimers.
2.1.1 7-oxanorbornenes (Class I)
2.1.2 dipyridylpyridazine (dpp) ligands (Class I)
2.2 bis-pyrimidines (Class II)
2.3.1 bis-(homo[n]azaladderenes) (Class II)
2.3.2 bis-([n]azaladderenes) (Class II)
2.4 bis-prolines
(Class III)
2.1.1 The 7-Oxa-benzonorbornene Chromophore
The use of Class I photodimerisation as a coupling technique can
be applied to different norbornene cyclobutene 1,2-diesters such
as 7-oxa benzonorbornadiene 17 (Scheme 6). Although Eberbach
has reported that both dimeric and rearranged products were obtained
from the rearrangement of simple fused cyclobutene-1,2-diesters
(Scheme 5), we have found that formation of side-reactions can
be effectively suppressed by using fused alicylics such as the
benzo-7-oxanorbornadiene 17. In this case, only a single
product 18 was observed.
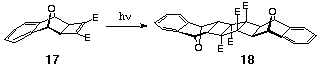
2.1.2 Di(2-Pyridyl)pyridazine Ligand Systems
This section gives an example where we have taken advantage of
the inverse electron-demand ability of 3,6-di(2-pyridyl)-s-tetrazine
(followed by DDQ oxidation to the dpp ligand) to react site specifically
at the norbornene p-bond of Smith's
diene without modifying the cyclobutene-1,2-diester group. This
constitutes an alternative route to functionalised cyclobutene-1,2-diesters
and has broad application. Irradiation of 19 furnishes
the dimer 20 in high yield (Scheme 7).
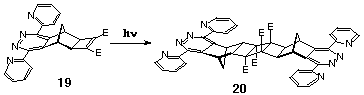
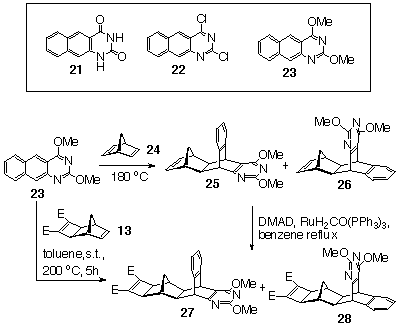
2.2 Synthesis of bis-pyrimidines
(CLASS II Dimerisations)
We have reported earlier on the synthesis of norbornene-fused pyrimidine building BLOCKS.[8] This approach involved the use of 2,4-dimethoxy-1,3-diazaanthracene 23, rather than the known 1,3diazaanthracen-2,4-dione 21[9] or 2,4-dichloro-1,3-diazaanthracene 22[9], as 23 alone in this series displays 1,3diene reactivity. The presence of the methoxy groups served to activate the 1,3diazaanthracene nucleus towards Diels-Alder cycloadditions and to provide the required Hbonding capacity of the uracil subunit in latent form.
2,4Dimethoxy-1,3-diazaanthracene (23) reacted thermally with
norbornadiene 24 to form the adducts 25 and 26.
A mixture of 25 and 26 was treated with DMAD/Ru(0)
under Mitsudo conditions to provide an isomeric mixture of the
adducts 27 and 28 (Scheme 8).[8]
Chromatographic separation on silica and subsequent fractional
crystallisation yielded pure adduct 27,[8]
whilst 28 (m.p. 124-126 oC) was isolated by reverse phase
preparative HPLC (C18 NOVA pack, methanol/water gradient elution
system (from 70/20 to 90/10 ratio over the period of 25 minutes,
flow rate 10 ml/min). Alternatively, a mixture of 27 and
28 was obtained by the Diels-Alder reaction of 1,3diazaanthracene
23 and Smith's diester 13 (Scheme 8).
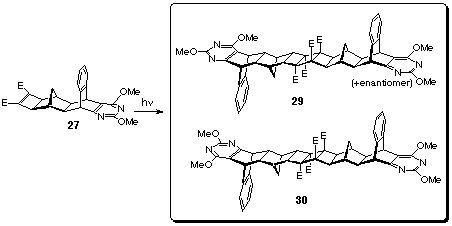
Enantiomeric mixtures of cyclobutene diesters 27 and 28
were separately dissolved in deuteriated chloroform and irradiated
at 254 nm wavelength for two hours in Rayonet photochemical mini-reactor
(Model RMR-600) (Scheme 9). The products were purified by radial
chromatography employing a gradient elution technique (started
with petroleum ether/ethyl acetate 3:1 mixture, ended with pure
ethyl acetate) and subsequent recrystallisation from dichloromethane
- methanol solvent mixture. White photodimers 29 and 30
(m.p. > 360 oC, 57% yield), 31 and 32 (m.p. >
360 oC, 61% yield) were obtained and these syn and meso
photodimers remained as mixtures as their separation was not
successful.
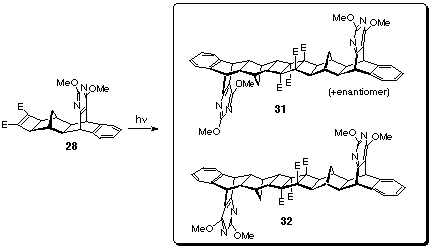
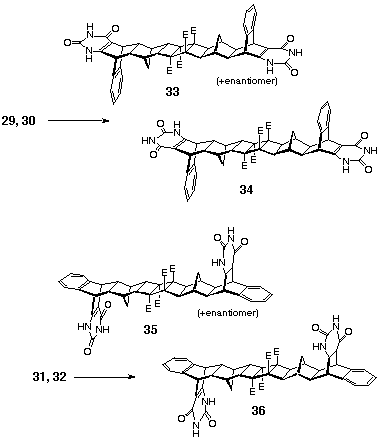
The tetramethoxy bis-pyrimidines 29-32 were transformed
to bis-uracils 33, 34 (m.p. > 380 oC, 80% yield),
35 and 36 (m.p. > 380 oC, 84% yield), Scheme
10, by treatment with hot hydrochloric acid (0.58 M, 100 oC, 30
hours). In these products, the signals for the protons of the
methoxy groups have disappeared as evident from the 1H
NMR (DMSO-d6) data and there are two
new resonances in the low-field region for the two non-equivalent
NH protons (d 10.86, 11.41 in 33,
34; d 10.98, 11.51 in 35, 36).
The methyl esters remained intact under these reaction conditions
as revealed by the resonances at d
3.70 and 3.71 in the 1H NMR (DMSO-d6)
spectra of 35, 36 isomeric product mixture. The corresponding
signals overlap and appear at d 3.65
in the bis-uracils 33, 34.
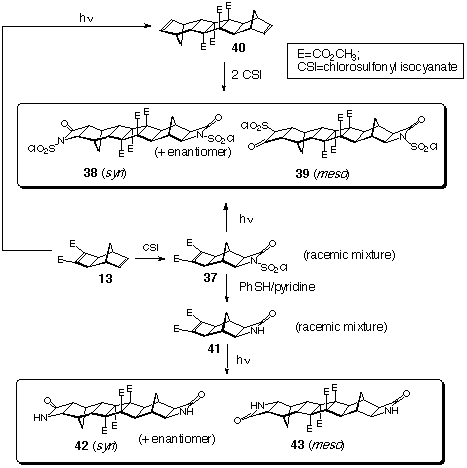
2.3 b-Lactam
containing Products
The synthesis of aza[n]ladderanes and azahomo[n]ladderanes possessing b-lactam functionality has been reported by us.[10] These rod-like aza[n]ladderanes and chevron-like azahomo[n]ladderanes structures have great potential, especially in regard to being latent bamino acids.
Bis-(N-chlorosulfonyl)azahomo[9]ladderanes, 38 and 39, and bis-(azahomo)-[9] ladderanes, 42 and 43, were synthesised via the steps indicated in Scheme 11. In both cases, the starting material was Smith's diene 13[11a] which has reacted with the chlorosulfonyl isocyanate site specifically at the norbornene p-bond to give the exo-adduct 37. A solution of 37 was irradiated (450W medium pressure mercury lamp, pyrex filter) to produce an inseparable mixture of bis-(N-chlorosulfonylazahomo)-[9]ladderans, 38 and 39 (m.p.>380oC), in 90% combined yield.
Another approach to the synthesis of 38 and 39, involved the photodimerisation of Smith's diester first. Diene 40[4a] was then treated with excess of chlorosulfonyl isocyanate to produce a similar mixture of bis-adducts 38 and 39 (not separated).
N-Chlorosulfonylazahomo)-[4]ladderane 37 was hydrolysed with thiophenol in pyridine to the b-lactam 41 which was photodimerised (CDCl3 solution, 450W medium pressure Hg lamp, pyrex filter) to form the bis-(b-lactam) structures 42 and 43 (m.p.>380oC), (Scheme 11).
This section of the work is by way of a preliminary account as separation of these different dimeric photoproducts has not been achieved. Accordingly, characterisation has been on mixtures. Spectral analysis has also been difficult since both the chlorosulfonyl derivatives and the free lactams each have very little difference between the individual isomers in the mixture. Further work is required to bring this section up to a more professioanl standard, and is presently being worked on.
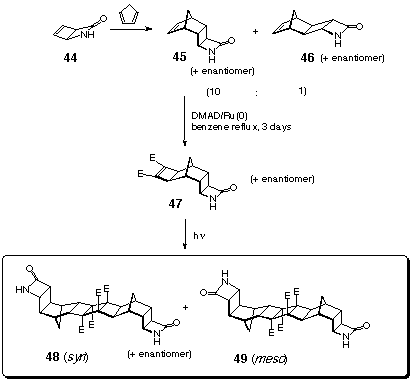
Bis-(azahomo)[11]ladderanes 48 and 49 (m.p. 258-260 oC) are one cyclobutane ring longer than 42 and 43.[10] They were prepared by the photodimerisation of an enantiomeric mixture of the diester-fused b-lactam compound 47 (CDCl3 solution, 450W medium pressure Hg lamp, pyrex filter, 0 oC), as outlined at Scheme 12. As expected, a mixture of syn- and meso-bis-adducts 48 and 49 was obtained.
The synthesis of the diester 47 was described[10]
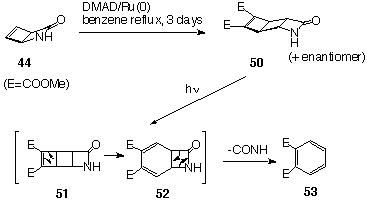
earlier, Scheme 12. It includes photoisomerisation of apyridone
to form aza[2]ladderane 44[12]
which further reacts with cyclopentadiene to produce an isomeric
mixture of norbornene-fused azaladderanes 45 and 46
in 10:1 ratio. 45 can be readily separated from this mixture
by fractional crystallisation. It underwent Ru(0) catalysed cycloaddition
of DMAD to yield 47.
The attempts to prepare a [7]diazaladderane by similar photodimerisation
of [3]azalladerane 45 failed. The only isolated product
was dimethyl phthalate 53 (Scheme 13). The proposed mechanism
of formation of 53 is outlined below.
2.4 Synthesis of bis-prolines
(CLASS III Dimerisation)
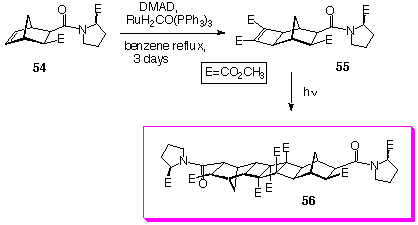
Although dimerisation reaction described so far, because of racemic mixtures used produced mixtures of dimers, we have shown that, if chiral starting material is used, such as compound 55 (prepared by Mitsudo reaction from compound 54)[15], only single product 56 was obtained (Scheme 14).
Conclusion. We have shown that photodimerisation of cyclobutene diesters can be effectively used ifor the preparation of large policyclic structres, bearing different effector groups attached at both ends.
Acknowledgements. The Australian Research Council is thanked for funding.
M. G. thanks the Centre for Molecular Architecture for the award
of a Ph D scholarship.
All computed structures were fully optimised by AM1 method[13].
Substrates are prepared as described in literature: 17,[5]
19,[14] 27 and 28,[8]
37, 41, 47[10]
and 54[15]. All new compounds
were fully characterised by spectroscopic techniques and their
molecular formula established by high resolution mass spectrometry.
Representative data follow:
Tetramethyl-25,26-dioxa-7,9,11,19,21,23-dodecadehydro-
1a,2b,3a,4b,5a,6b,13b,14a,15b,16a,17b,18a-
octacyclo[16.6.1.16,1302,1703,1604,1505,1407,12019,24]hexacosa-3,4,15,16-
tetracarboxylate
(18).
The solution of 17 (100 mg) in CDCl3 (1.0 ml), in a quartz NMR tube was irradiated at 300 nm in Rayonet reactor (Model RMR-600). The progress of reaction was followed by NMR, yielding a quantitative conversion to product within 4 hours. The precipitate was formed and separated by filtration to afford a colourless solid, 65 mg (65%), m. p. 330-332 oC.
1H NMR (300 MHz, CDCl3) d
2.68 (4H, s); 3.86 (12H, s); 5.5 (4H, s); 7.21-7.29 (8H, m); 13C
NMR (300 MHz, CDCl3) d 44.2; 51.9;
55.9; 80.6; 119.9; 127.4; 143.2; 168.2; HRMS for C32H28O10
requires for M+ 572.1687, found 572.1682.
Tetramethyl-8,11,20,23-tetra(2'-pyridyl)-9,10,21,22-tetraaza-7,9,11,19,
21,23-dodecadehydro-1a,2b,3a,4b,5a,6b,13b,14a,15b,16a,17b,18a-octacyclo
[16.6.1.16,1302,1703,1604,1505,1407,12
019,24]hexacosa-3,4,15,16-tetracarboxylate (20).
A solution of 19 (60 mg) in CDCl3 (0.5 ml), in a quartz NMR tube was irradiated at 300 nm in Rayonet photoreactor (Model RMR-600) for 2 hours. Evaporation of solvent in vacuo yielded a yellow coloured solid. Product was purified by radial chromatography (petroleum ether : ethyl acetate 10:1, then the solvent was gradually increased to ethyl acetate) to afford a colourless solid (48 mg, 80%, m.p. 344-345 oC).
1H NMR (300 MHz, CDCl3) d
1.54( 2H, d, J=11.34 Hz); 2.20 (2H, d, J=11.1 Hz);
2.72 (4H, s); 3.72 (12H, s); 4.52 (4H, s); 7.34-7.35 (4H, m);
7.87 (4H, t, J=7.11 Hz); 8.46 (4H, d, J=7.41 Hz);
8.63 (4H, s); 13C NMR (300 MHz, CDCl3) d
41.5; 42.7; 45.6; 51.9; 57.0; 123.1; 124.3; 137.2; 147.1; 149.5;
153.1; 155.4; 165.7; HRMS for C50H40O8N8requires for
M+ 880.2968, found 880.2969.
(5b,5ab,6a,9a,9ab,10b)-6a,8a[3',4']Cyclobuta-7,8-dicarbomethoxy-2,4-dimethoxy- 5,5a,6,6a,8a,9,9a,10-octahydro-5,10[1",2"]-benzeno-6,9-methanobenzo[g]quinazoline (28)
Procedure A: A mixture of adducts 25 and 26 (200 mg, 0.60 mmol) was refluxed in benzene with DMAD (75 ml, 0.60 mmol) and RuH2CO(PPh3)3 catalyst (30 mg, 0.03 mmol) for 3 days. After radial chromatographic separation of the reaction mixture, products 27 and 28 (210 mg of crude product, 73%) and some starting material were isolated. Isomer 27 was isolated in a pure form (m.p. 224-225 oC) by fraction crystallisation (impurities and the other isomer remain in petroleum ether), followed by precrystalisation from ethyl acetate - petroleum ether.[8] The other isomer (28, m.p. 124-126 oC) remained in the mixture as an oil and was isolated by utilising preparative HPLC technique. C18 NOVA pack, reverse phase column was used, methanol/water gradient elution system (from 70/20 to 90/10 ratio over the period of 25 minutes, flow rate 10 ml/min).
Procedure B: 2,4-Dimethoxy-1,3-diazaanthracene (23) (100 mg, 0.42 mmol), Smith's diester (2.25 g, 9.6 mmol) and aniline (1 drop) were heated at 200 oC in a sealed vessel and inert atmosphere for 5.5 hours. The products 27 and 28 were isolated by chromatogaphy, first on the silica column (ethyl acetate:petroleum ether 1:1 eluent) as the second fraction, then by radial chromatography (10% acetic acid in ethyl acetate:petroleum ether 1:6 solvent mixture) as the fourth component (second that was black under 254 nm wavelength). 69 mg (35 % yield) of the mixture of isomers was obtained. The separation of two isomers was as same as described above.
1H NMR (300 MHz, CDCl3)
d 0.06 (1H, d, J=12.5 Hz),
0.77 (1H, d, J=12.5 Hz), 1.83 (2H, narrow m), 2.19 (1H,
s), 2.28 (1H, s), 2.57 (1H, s), 2.59 (1H, s), 3.74 (3H, s), 3.75
(3H, s), 3.96 (3H, s), 3.97 (3H, s), 4.32 (1H, d, J=2.2
Hz), 4.59 (1H, d, J=2.5 Hz), 7.09 (2H, m), 7.27 (2H, m);
1C NMR (75 MHz, CDCl3)
d 25.3, 37.1, 37.2, 39.6, 47.9, 48.0
(overlap), 48.9, 50.0, 51.6, 51.7, 53.6, 54.5, 112.9, 123.0, 124.0,
125.8, 126.0, 141.0, 141.3, 142.3, 143.6, 161.06, 161.14, 161.2,
164.6, 173.3; HRMS for C27H26N2O6
requires for M+ (m/e) 474.1791.
Found: 474.1784.
Pyrimidine photodimers 29-32
Photodimerisation procedure: Diesters 27 and 28 were placed in a quartz NMR tubes (F 5 mm), dissolved in CDCl3 and irradiated at 254 nm wavelength in a Rayonet photochemical mini-reactor (Model RMR-600). The conversion to photodimers was followed by the means of 1H NMR, and was completed after about 2 hours. A small amount of polymeric material was formed and removed on the silica column. Purified products were further recrystallised from dichloromethane - methanol solvent mixture to yield white powders of photodimers. The syn and meso photodimers remained as a mixtures as their separation was not successful.
Photodimers 29 and 30
yield 57%, m.p. >360 oC; 1H NMR (300 MHz, CDCl3) d -0.44 (2H, d, J=12.2 Hz), 0.96 (2H, d, J=12.2 Hz), 1.79 (4H, m), 2.06 (4H, narrow m), 2.23 (4H, brs), 3.65 (3H, s), 3.65 (6H, s), 3.66 (3H, s), 3.92 (6H, s), 3.93 (6H, s), 4.21 (2H, brs), 4.44 (2H, brs), 7.14 (8H, m); 1C NMR (75 MHz, CDCl3/CD3OD) d 27.0, 39.1, 41.2, 41.5, 45.1, 46.48, 46.5 (overlap), 48.8, 50.7 (overlap), 53.0, 53.9, 56.3 (overlap), 114.3, 123.7, 124.6, 126.0, 126.3, 138.7, 140.5, 162.4, 163.8 (C4, C9), 167.8, 167.9, 174.6. HRMS (positive ES) for C54H53N4O12 requires for M++H (m/e) 949.3660. Found: 949.3701.
Photodimers 31 and 32
yield 61%, m.p. >360 oC; 1H NMR (400 MHz,
CDCl3/CD3OD) d
0.14 (2H, d, J=12.5 Hz), 1.22 (2H, d, J=12.5 Hz),
1.81 (4H, narrow m), 2.18 (2H, brs), 2.21 (2H, brs), 2.34 (4H,
brs), 3.68 (6H, s), 3.70 (6H, s), 3.95 (6H, s), 3.96 (6H, s),
4.19 (2H, d, J=2.1 Hz), 4.52 (2H, d, J= 2.6 Hz),
7.09 (4H, m), 7.21 (4H, m); 1C NMR
(100 MHz, CDCl3/CD3OD)
d 28.4, 39.1, 41.6, 41.7, 46.6, 46.7,
46.7, 47.7, 47.8, 49.8, 51.2 (overlap), 53.4, 54.3, 56.5, 56.7,
112.6, 122.8, 123.7, 125.8, 126.1, 141.5, 143.0, 163.1, 164.4,
167.9, 167.9, 168.1, 168.2, 173.0.
HRMS (positive ES) for C54H53N4O12
requires for M++H(m/e) 949.3660.
Found: 949.3581.
Bis-uracil photodimers 33-36
Hydrolysis procedure: The tetramethoxy bis-pyrimidines
29-32 were hydrolysed to bis-uracils 33-36
by treatment with hot hydrochloric acid (0.58 M, 100 oC, 30 hours).
The hydrochloric acid solution was evaporated in vacuo, and the
residual white solid dissolved in a mixture of methanol and sodium
hydroxide (2M). Upon standing at room temperature overnight,
white crystals of the products were obtained. The syn
and meso photodimers are characterised as mixtures.
Bis-uracil photodimers 33 and 34
yield 80%, m.p. >380 oC; 1H NMR (300 MHz, DMSO-d6) d -0.72 (2H, d, J=11.9 Hz), 0.89 (2H, d, J=11.9 Hz), 1.77 (2H, d, J=7.7 Hz), 1.89 (2H, d, J=7.7, 1.6 Hz), 1.98 (2H, brs), 2.03 (2H, brs), 2.25 (4H, brs), 3.65 (12H, s), 4.17 (2H, brs), 4.36 (2H, brs), 7.26 (4H, m), 7.29 (4H, m), 10.86 (2H, s), 11.41 (2H, s).
Bis-uracil photodimers 35 and 36
yield 84%, m.p. >380 oC; 1H NMR (300 MHz,
DMSO-d6) d
1.09 (2H, d, J=11.2 Hz), 1.47 (2H, d, J=11.2 Hz),
1.68 (2H, dd, J=7.9, 1.6 Hz), 1.79 (2H, dd, J=7.9,
1.6 Hz), 2.16 (2H, brs), 2.19 (2H, brs), 2.28 (2H, brs), 2.30
(2H, brs), 3.70 (6H, s), 3.71 (6H, s), 4.20 (2H, brs), 4.43 (2H,
brs), 7.19 (4H, m), 7.32 (4H, m), 10.98 (2H, s), 11.51 (2H, s).
6,17-bis(chlorosulfonyl)-tetramethyl-6,17-diazanonacyclo
1b,2a,3b,4a,5b,8b,9a,10b,11a,12b,13a,14b,15a,18a,19b,20a
[10.8.6.14,9114,1901,1202,1103,1005,8015,18]docosa-7,16-dione-1,2,11,12-
tetracarboxylate
(38, 39).
A solution of N-chlorosulfonyl substituted b-lactam 37 (50 mg, 0.133 mmol) in a Pyrex NMR tube in chloroform-d (0.5 ml) was irradiated for 25 min using a 450 W medium pressure Hg lamp. Evaporation of the solvent in vacuo gave a colourless solid (45 mg, 90%, m.p. >380 oC).
1H-NMR (300 MHz, CDCl3) d
1.75 (2H, d, J=13.3 Hz); 2.17 (2H, d, J=13.3 Hz);
2.44 (4H, m); 2.81 (2H, s); 3.00 (2H, s); 3.25 (2H, d, J=4.3
Hz); 3.78 (6H, s); 3.83 (6H, s); 4.08 (2H, d, J=4.3 Hz);
13C-NMR (300 MHz, CDCl3) d
27.0; 38.5; 40.0; 41.4; 43.3; 52.2; 52.3; 56.9; 57.0; 57.0;
59.8; 160.9; 166.9; 166.9; IR (KBr/cm-1): 1815.3 s; 1747.8 s;
1727.3 s; 1321.4 s; 1269.3 s; MS (m/e, %); 149 (10); 80 (14);
66 (25); 64 (100); 44 (71).
Tetramethyl-6,17-diazanonacyclo 1b,2a,3b,4a,5b,8b,9a,10b,11a,12b,13a,14b,15a,18a,19b,20a [10.8.6.14,9114,1901,1202,1103,1005,8 015,18] docosa-7,16-dione-1,2,11,12-tetracarboxylate (42, 43).
A solution of b-lactam 41 (50 mg, 0.18 mmol) in Pyrex NMR tube in CDCl3 (0.5 ml) was irradiated for 30 min using a 450 W medium pressure Hg lamp. Evaporation of solvent in vacuo gave a colourless solid (45 mg, 90%), m.p. >350 oC.
1H-NMR (300 MHz, CDCl3) d
1.68 (2H, d, J=12.4 Hz); 1.96 (2H, d, J=12.4
Hz); 2.36 (4H, m); 2.59 (2H, d, J=9.5 Hz); 3.01 (2H, s);
3.40 (2H, t, J=3.4 Hz); 3.80 (3H, s); 3.81 (3H, s); 3.82
(3H, s); 3.83 (3H, s); 6.14 (2H, s); 13C-NMR
(300 MHz, CDCl3) d 25.8; 36.4; 40.6;
42.1; 43.4; 43.6; 51.0; 51.7; 56.6; 57.1; 57.2; 54.2; 57.6; 167.5;
167.6; 167.7; 167.8; 168.2; IR (KBr/cm-1): 3447.4 s; 2954.9 m;
1742.2 s; 1654.1 w; 1269.0 s; MS (m/e, %); 523 (M-OMe, 0.3); 279
(45); 167 (19); 149 (52); 70 (100); 66 (88); HRMS for C28H30O10N2
requires for M+ 554.1900, found 554.1917.
Tetramethyl-9,22-diazaundecacyclo[16.6.1.16,1302,1703,1604,1505,14 07,1208,11019,24 020,23]hexacos-5,6-dione-3,4,15,16-tetracarboxylate (48, 49).
A solution of 47 (50 mg) in CDCl3, in a Pyrex NMR tube was irradiated at 300 nm in a Rayonet mini-reactor, Model RMR-600. The progress of reaction was followed by NMR, yielding a quantitative conversion to product within 8 hours. The precipitate formed was separated by filtration to afford a colourless solid, 35 mg (70%), m. p. >340 oC.
1H NMR (400 MHz, CDCl3) d
1.36 (2H, d, J=10.6 Hz); 2.11-2.13 (2H, m); 2.22 (2H, d,
J=10.8 Hz); 2.36-2.50 (2H, m); 2.56-2.57 (2H, m); 2.60-2.61
(2H. m); 2.68-2.73 (2H, m); 2.84 - 2.89 (2H, m); 3.48 (2H, br
s); 3.78 (6H, s); 3.80 (6H, s); 3.95 (2H, br s); 5.84 (2H, br
s); 13C NMR (400 MHz, CDCl3) d
37.7; 39.9; 40.9; 40.9; 41.2; 41.3; 49.2; 50.6; 51.0; 52.0; 52.3;
53.8; 57.5; 168.2; 168.3; 171.9; MS (m/e, %); 278 (36); 277 (100):
199 (24); 117 (36); 77 (52); HRMS for C32H34O10N2 requires
for M+ 606.2210, found 606.2213.
L-Proline,1-[[3',4',8'-tris(methoxycarbonyl)-1'a,2'b,5'b,6'a,7'b,8'b-tricyclo [4.2.1.02',5'] non-3'-en-7'-yl]carbonyl]-, methyl ester, [6'R- (exo, exo)] (55).
A mixture of amide 54 (3.75 g, 12.24 mmol), RuH2CO(PPh3)3 catalyst (0.5 g, 0.58 mmol), DMAD (8.64 g, 61.2 mmol) in dry benzene (20 ml) was refluxed at 70 oC for 3 days, under an nitrogen atmosphere. The solvent was then removed in vacuo to afford a black coloured oily residue. The mixture was then subjected to chromatographi separation (column, silicagel, starting eluent petroleum ether : ethyl acetate 2:1, then the solvent polarity was gradually increased to ethyl acetate) to afford brown coloured oil (6.18 g). The product was subjected to another chromatography (column, silicagel, starting eluent petroleum ether : ethyl acetate 1:1, then the solvent polarity was gradually increased to ethyl acetate) to afford a yellow coloured oil (4.84 g, 88%).
1H-NMR (300 MHz, CDCl3) d
1.32 (2H, d, J=11.7 Hz); 1.88-1.96 (3H, m); 2.49-2.51
(2H, m); 2.63-2.68 (5H, m); 3.52 (3H, s); 3.53-3.63 (2H, m); 3.63
(3H, s); 3.69 (3H, s); 3.71 (3H, s); 4.28-4.32 (1H, m); 1C-NMR
(300 MHz, CDCl3) d 24.7; 27.9; 29.0;
35.7; 36.3; 38.3; 46.0; 46.4; 47.2; 47.9; 49.4; 51.7; 51.9; 58.6;
60.1; 141.6; 141,7; 160.9; 160.9; 170.8; 170.8; 172.2; 172.6;
MS (m/e, %): 449 (M, 0.5); 322 (18); 321 (100); 201 (17); 242
(33); 70 (61); 59 (28); HRMS for C22H27O9N requires for
M+ 449.1685, found 449.1686; [a]D20=-34.5
o, (c=1.0, CHCl3).
bis-(L-Proline,1)-[[1',2',6',9',10',13'-hexa(methoxycarbonyl)-
1'b,2'a,3'b,4'a,5'b,6'b,7'a,8'b,9'a,10'b,11'a,12'b,13'a,14'a,15'b,16'a,-
] heptacyclo[8.6.0.14',7'112',15'01',10'02',9'011',16']octadec-5',13'-di-yl]carbonyl]-
methyl
ester, [4'R, 12'R- (exo, exo)] (56).
A solution of ester 55 (20 mg, 0.045 mmol) in chloroform (0.5 ml), in a quartz NMR tube was irradiated in a Rayonet photochemical mini reactor (Model RMR-600, 253 nm) for 2 hours. The reaction was followed by proton NMR. Evaporation of solvent in vacuo gave a colourless solid (18 mg, 96.0%, m.p. >330 o, dec.).
1H-NMR (300 MHz, CDCl3) d
1.81 - 2.18 (6H, m); 2.23 (2H, d, J=12.2 Hz); 2.46 (6H,
s), 2.59 - 2.71 (10H, m); 3,47 (6H, s); 3.70 (6H, s); 3.71 - 3.73
(4H, m); 3.76 (6H, s); 3.77 (6H, s); 4.30 - 4.31 (2H, m); 13C-NMR
(300 MHz, CDCl3) d 20.9; 24.9; 29.3;
41.2; 42.7; 44.9; 45.3; 47.4; 47.7; 49.5; 51.7; 51.8; 51.9; 52.2;
57.0; 58.8; 60.3; 167.9; 167.7; 170.9; 171.9; 172.9; MS (m/e,
%): 898 (M+, 9); 868 (9); 839 (12) 770 (100), 592 (40); 242 (41);
[a]D20=-23.0 o, (c=0.2, CHCl3).
Details about different coupling
methods
ACE Coupling[16]
In the ACE coupling technique, the ester-activated cyclobutene
epoxide ring opens to a 1,3dipolar species, which is trapped
by ring-strained alicyclic dipolarophiles. It was found that
fused norbornene, 7-oxanorbornene and benzonorbornadienes can
act as one building BLOCK (A-BLOCK) and fused cyclobutene
epoxide as the other (B-BLOCK). Heating of two BLOCKs together
gives the linear (exo,exo-coupled) product exclusively
if fused norbornenes are used as the A-BLOCK (Scheme 15).
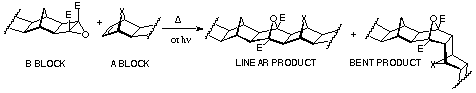
Advantages: 1) applicable to all compounds possessing norbornene p-bond; 2) simple procedure to make epoxides; 3) both homo- and hetero-coupled products can be prepared; 4) mild conditions if photo-ACE is used; 5) a variety of solvents can be used.
Disadvantages: 1) not all norbornene-fused compounds undergo
Mitsudo/epoxidation reaction sequence in the precoupling step;
2) thermal unstability of some dipolarophile component at the
reaction temperatures, typically 140 oC; 3) 7-oxa and 7-isopropylidene
norbornene dipolarophiles give mixtures of linear and bent products.
Aza-ACE Coupling[17]
The Aza-ACE coupling technique resembles ACE coupling
method, except that it produces a 7-azanorbornene at the coupling
site. Ester-activated aziridino cyclobutanes (B-BLOCK)
open to a 1,3-dipole, which is readily trapped by ring-strained
alicyclic dipolarophiles (A-BLOCK). Regardless of which
A-BLOCK is used, only linear products are obtained (Scheme
16).
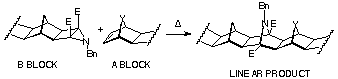
Advatages: 1) applicable to all compounds possessing norbornene p-bond; 2) both homo- and hetero-coupled products can be prepared; 3) high stereoselectivity - only linear products are formed.
Disadvantages: 1) not all norbornene-fused compounds undergo
Mitsudo/benzyl azide reaction sequence in the precoupling step;
2) azide can react on the unsubstituted norbornene double bond;
3) thermal instability of some dipolarophile components at the
reaction temperatures, eg 80 oC.
The thermal coupling of alkenes by reaction with certain 1,3,4-oxadiazoles
(OD) provides 7-oxanorbornanes in which the C2,3 and C5,6 components
are derived from alkenes and the 1,3,4-oxadiazole contributes
the C1,4 bridgehead carbons and the oxa-bridge (Scheme 17).
The geometry of the products can be altered by choosing different
atoms and groups of the norbornene bridge X in A-BLOCK.
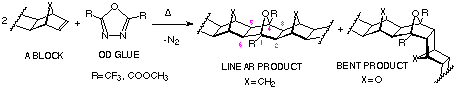
Advantages: 1) applicable to all compounds possessing norbornene p-bond; 2) only homo-coupled products can be prepared.
Disadvantages: 1) thermal instability of some alkene substrates
- reaction conditions 140 oC; 2) nonstereospecific - mixtures
of products are often produced.
s-Tetrazine
Coupling[19-21]
The use of 3,6-bis(2'pyridyl)-s-tetrazine (s-tetrazine) as a coupling agent is another method for joining fused norbornenes and 7-oxanorbornenes (A-BLOCKS), Scheme 18. The reaction is stereoselective and produces extended-frame adducts only (exo, exo-addition). Being a two-step process, this coupling technique allows construction of homo- (if the same fused norbornene is coupled) or hetero-coupled products (if different fused norbornenes used). It is critical to include triethylamine in the reaction mixture to avoid isomerisation of the intermediate dihydropyridazine.
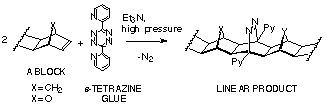
Advantages: 1) applicable to all compounds possessing norbornene p-bond; 2) both homo- and hetero-coupled products can be prepared; 3) stereospecific - extended-frame products are produced exclusively; 4) mild reaction conditions, eg mainly room temperature and high pressure.
Disadvantages: 1) requires high pressure conditions; 2)
limited solvent choice; 3) produces retro- Diels-Alder products
in some cases.
The 1,2,4-triazine coupling method is an analogue of the s-terazine coupling method. It is used to join fused norbornenes and 7-oxanorbornenes (A-BLOCKS), Scheme 19. The reaction is stereoselective and the extended-frame adducts are formed exclusively.
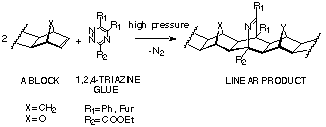
Advantages: 1) applicable to all compounds possessing norbornene p-bond; 2) both homo- and hetero-coupled products can be prepared; 3) stereospecific - linear products are generated exclusively; 4) mild reaction conditions, eg room temperature and high pressure.
Disadvantages: 1) requires high pressure conditions; 2)
limited solvent choice; 3) produces retro- Diels-Alder products
in some cases.
[1] Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett, 1998, 566.
[2] Warrener, R. N. Chem. Aust., 1992, 59, 578.
[3] (a) Spanget-Larsen, J. Gleiter, R. Tetrahedron 1983, 3345; (b) Rondan, N. G.; Paddon-Row, M. N.; Caramella, P.; Mareda, P.; Mueller, P.; Houk, K. N. J. Am. Chem. Soc. 1982, 104, 4974 and references cited therein.
[4] (a) Eberbach, W. Chem. Ber. 1974, 107, 3287; (b) Warrener, R. N.; Groundwater, P.; Pitt, I. G.; Butler, D. N.; Russell, R. A. Tetrahedron Lett. 1991, 32, 1885; (c) Butler, D. N.; Russell, R. A.; Waring, R. B.; Warrener, R. N. Aust. J. Chem. 1984, 37, 1293.
[5] (a) Mitsudo, T.; Kokuryo, K.; Shinsugi, T.; Nakagawa, Y.; Watanabe, Y.; Takegami, Y. J. Org. Chem. 1979, 44, 4492; (b) Mitsudo, T.; Naruse, H.; Kondo, T.; Ozaki, Y.; Watanabe, Y. Angew. Chem., Int. Ed. 1994, 33, 580.
[6] Snider, B. R.; Rousch, D. M.; Rodini, D. J.; Gonzalez, D.; Spindell, D. J. Org. Chem. 1980, 45, 2773; Snider, B. R.; Rodini, D. J.; Conn, R. S. E.; Sealfon, S. J. Am. Chem. Soc. 1979, 101, 5283.
[7] (a) Seebach, D. Ber. 1964, 97 , 2953; (b) Trecker, D. J. Organic Photochemistry, Vol. 2, Chapman, O. L. Ed.; Marcel Dekker, New York, 1969, pp. 63116.
[8] Warrener, R. N.; Golic, M.; Butler, D. N. Synlett 1997, 1105.
[9] Sengupta, S. K.; Chatterjee, S.; Prototapa, H. K.; Modest, E. J. J. Org. Chem. 1972, 37, 1323.
[10] Warrener, R. N.; Russell, R. A.; Margetic, D. Synlett 1997, 38.
[11] (a) Smith, C. D. J. Am. Chem. Soc. 1966, 88, 4273; (b) Warrener, R. N.; Butler, D. N. Aldrichimica Acta 1997, 30, 119.
[12] Corey, E. J.; Streuth, J. J. Am. Chem. Soc. 1964, 86, 950.
[13] Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F. J. Am. Chem. Soc. 1985, 107, 3902.
[14] Warrener, R. N.; Elsey, G. M.; Sankar, I. V.; Butler, D. N.; Pekos, P.; Kennard, C. H. L. Tetrahedron Lett. 1994, 35, 6745.
[15] North, M.; Zagotto, G. Synlett, 1997, 38.
[16] Warrener, R. N.; Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A. Chem.Commun. 1997, 1023.
[17] Butler, D. N.; Malpass, J. R.; Margetic, D.; Russell, R. A.; Sun, G.; Warrener, R. N. Synlett 1998, 588.
[18] (a) Vasiliev, N. V.; Lyashenko, Y. E.; Kolomietz, A. F.; Sokolskii, G. A. Khim. Geterotsikl Soedin. 1987, 562; (b) Vasiliev, N. V.; Lyashenko, Y. E.; Patalakha, A. E.; Sokolskii, G. A. J. Fluorine Chem. 1993, 65, 227; (c) Seitz, V. G.; Wassmuth, H. Chemiker Zeitung 1988, 112, 80; (d) Seitz, G.; Gerninghaus, C. H.; Pharmazie 1994, 49, 102; (e) Thalhammer, F.; Wallfahrer, U.; Sauer, J. Tetrahedron Lett. 1988, 29, 3231; (f) Warrener, R. N.; Margetic, D.; Tiekink, E. R. T.; Russell, R. A. Synlett. 1997, 196.
[19] (a) Dittmar, W.; Heinrichs, G.; Steigel, A.; Troll, T.; Sauer, J. Tetrahedron Lett. 1970, 1623; (b) Heinrichs, G.; Krapf, H.; Schroder, B.; Steigel, A.; Troll, T.; Sauer, J. Tetrahedron Lett. 1970, 19, 1617.
[20] (a) Elix, J. A.; Wilson, W. S.; Warrener, R. N. Syn. Commun. 1972, 2, 73; (b) Elix, J. A.; Wilson, W. S.; Warrener, R. N.; Calder, I. C. Aust. J. Chem. 1972, 25, 865; (c) Wilson, W. S.; Warrener, R. N. Tetrahedron Lett. 1970, 55, 4787.
[21] Warrener, R. N.; Margetic, D.; Russell, R. A. Synlett. 1998, 585.