| [Related articles/posters: 111 091 121 ] |
Introduction
Central (narcotic) analgesics present a very important class of drugs, widely used in surgical procedures, in treatment of general postoperative pain, in cancer pain and other conditions [1,2].
The 4-anilidopiperidines are the most potent class of central analgesics known to date [1,2]. The prototype of this class, fentanyl 1, is about 300 times more potent than morphine in mice and rats [3], compared to 50-100 times in humans. A very large number of fentanyl analogues have been synthesized since 1964, including acyclic compounds like diampromide 2, and 2,3-seco-fentanyl 3, Fig.1 [4]. The corresponding pharmacological
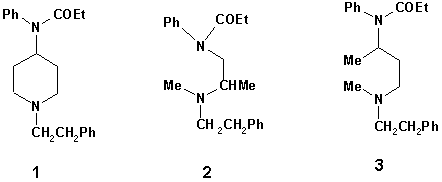
Figure 1
data were published, and the structure-activity relationship, (SAR), for this class of central analgesic were examined in some detail [1-3,5,6]. The SAR studies revealed, among other factors, a great influence of the stereochemistry upon the analgesic activity.
As a part of our ongoing efforts towards the preparation of novel active analogues of fentanyl herein we wish to report theoretical studies and preliminary pharmacological examination of acyclic analogs ( 2 and 3, Fig.1) with analgesic activity.
Experimental
The 2,3-seco-fentanyl has been prepared [4] in our laboratory and was examined for analgesic activity by using rat-tail immersion test and the mouse hot plate test. The preliminary values of the relative order of potency are reported in Table 2.
Computational Method
The computational method used for the study of the conformational space of the molecules was the MC-MM2 method [7]. The method is based on Allinger‚s MM2 program [8] and supplemented with Monte Carlo type subroutine for generation of the new conformations using random variation of the randomly selected torsional angles. The neutral form of the compounds have been used, with dielectric constant 1.5 applied in the calculations of the electrostatic interactions.
The molecular electrostatic potentials were calculated using AM1 semiempirical method implemented to HyperChem 4.0 program [9]. The QSAR properties, reported in Table 2 were calculated using ChemPlus program [9]. The Overlay option of this program have been used to locate fentanyl like conformations of 2 and 3. The water accessible surface areas and the related octanol/water free energies of transfer were calculated by PCMODEL program [10].
Results and Discussion
The great influence of stereochemistry of the fentanyl class of compounds upon their analgesic activities is very well known. The theoretical studies of the active analogs of fentanyl [11] led to the proposal of some elements of pharmacophore necessary for optimum interaction with the receptor. These are:
- piperidine ring in the chair conformation
- N-phenethyl and 4-N-phenylpropanamide substituents trans and both equatorial
- trans configuration of the amide group
- extended conformation of the N-phenethyl substituent
- nearly perpendicular orientation of the aromatic ring (N-Ph) with respect to the amide function.
Also the role of the four structure elements, necessary for the optimum receptor recognition, has been postulated [11]. These are:
- protonated amine nitrogen
- polar function (C=O) capable of hydrogen bonding with a receptor
- two aromatic rings
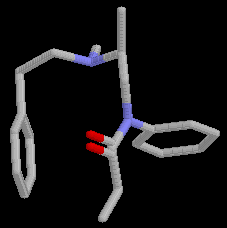
During the conformational search calculations at least three runs with 1500 steps each, were done for each of the compounds 2 and 3, using different conformations as the starting point. As expected the compounds were highly flexible. More than 50 conformations were located in the low energy conformational region. The global minimum conformations of 2 (E= 8.08 kcal/mol), Fig.2, and 3 ( E= 5.71 kcal/mol), Fig.3 differ considerably from the proposed [11] receptor recognized conformation of fentanyl, Fig.4.
 .
.
Figure 2. Global minimum conformation of 2.
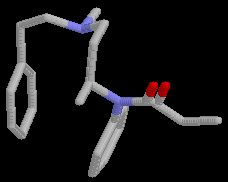
Figure 3. Global minimum congormation of 3.
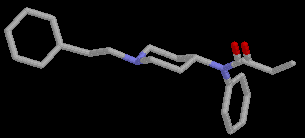
Figure 4. Postulated receptor recognized conformation of 1.
The structures of 2 and 3, corresponding to the proposed fentanyl class pharmacophore have been found among the high energy conformations. The overlap of the postulated receptor recognized conformation of 2 (E= 12.76 kcal/mol), and 3 (E= 16.84 kcal/mol) with fentanyl is presented in Fig.5 and Fig.6, respectively. Compared to 3, the receptor recognized conformation of 2 is lower in energy, relative to its global minimum conformation, but its overlap with fentanyl is not so good.

Figure 5. Overlap of postulated receptor recognized conformations of 1 and 2.
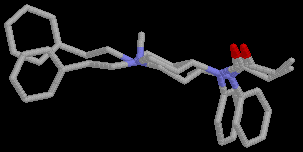
Figure 6. Overlap of postulated receptor recognized conformations 1 and 3.
.
Table 1.
Molecular Properties of 1, 2 and 3 in their Postulated Receptor Recognized Conformations|
Compound |
SWa |
Sb |
D Gc |
SUd |
Ve |
HEf |
LogP |
Rg |
Ph |
|
Fentanyl 1 |
587 |
375 |
2.3 |
627 |
1075 |
-1.25 |
4.25 |
103.8 |
40.5 |
|
Diampromide 2 |
591 |
391 |
2.9 |
604 |
1050 |
-0.75 |
4.66 |
100.8 |
39.4 |
|
2,3-seco-Fentanyl 3 |
601 |
395 |
2.8 |
642 |
1111 |
-0.68 |
4.72 |
105.6 |
41.2 |
a
Water accessible surface (A2), b VDW surface (A2), c Octanol-water free enrgy of transfer (kcal/mol) : PCMODEL; d Surface area (grid) (A2), e Volume (A3), f Hydration energy (kcal/mol), g Refractivity (A2), h Polarizability (A3) : ChemPlus.The calculated 3D electrostatic potentials of the three compounds 1, 2 and 3 are similar and so are the other molecular properties reported in Table 1, even though fentanyl appears to be somewhat more hydrophilic compared to 2 and 3.
Table 2.
Relative Analgesic Potencies|
Compound |
Relative Potency |
|
Fentanyl 1 |
1 |
|
Diampromide 2 |
0.006 a |
|
2,3-seco-Fentanyl 3 |
0.01 - 0.05 |
a
Ref. 1.Therefore we believe that the reduced analgesic potencies of 2 and 3, Table 2, are most likely related to their high flexibility, and the high energy of their receptor recognized conformations.
References
[1
] A. F. Casy, R. T. Parfitt "Opioid Analgesics", Plenum Press, N.Y., USA, 1986[2] A. F. Casy "Opioid Receptors and Their Ligands", p.178-272 in Advances in Drug Research, Ed. B. Testa, Vol 18, Academic Pres, London, 1989
[3] P. G. H. Van Daele, M. F. De Bruyn, J. M. Boey, S. Sanczuk, J. T. M. Agten, P. A. J. Janssen; Arzneim.-Forsch. (Drug Res.) 1976, 26, Nr. 8, 1521
[4] M. Ivanovic, I.V. Micovic, unpublished results ( part of the PhD thesis of M.Ivanovic).
[5] A. F. Casy, F.O. Ogungbamila; J. Pharm. Pharmacol. 34, 210. (1982); Eur. J. Med. Chem. 1983, 18, 56
[6] P. A. J. Janssen, C. A. M. van der Eycken in "Drugs Affecting the CNS" (A. Burger,ed.), Vol.2, s. 25, Dekker, N. Y. 1968
[7] Lj. Dosen-Micovic, Tetrahedron , 1995, 51, 6789.
[8] N.L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127.
[9] Hypercube,Inc. 419 Phillip St., Waterloo, ON N2L 3X2, Canada.
[10] Serena Software, Box 3076, Bloomington, IN 47402-3076, J.J. Gajewski, K.E.
Gilbert, J. McKelvey, Adv. Mol. Mod.,1990, 2, 65.
[11] C. Cometta-Morini, P.A. Maguire, G. D. Loewe, Mol. Pharmacol., 1992, 41 185; Lj. Dosen-Micovic, M. Ivanovic, G. Roglic, I.V. Micovic, J. Serb. Chem. Soc., 1996, 61, 1075; Lj. Dosen-Micovic, G. Roglic, I.V. Micovic, M. Ivanivic, Electronic Journal of Theoretical Chemistry, 1996, 1, 199