| [Related articles/posters: 028 026 052 ] |
 Heterocyclic Synthesis via Domino Cascade Processes Heterocyclic Synthesis via Domino Cascade Processes
Albert
Padwa |
|---|
In recent years, consecutive pericyclic reactions involving at least
one cycloaddition have been utilized for the synthesis of complex polycyclic
ring systems [1]. In the realm of synthesis in which a premium is put on
the rapid construction of polyfunctional, highly bridged carbon and heteroatom
networks, the [4+2]-cycloaddition has emerged as one of the foremost synthetic
methods [2]. Well known and extensively studied for many decades, the Diels-Alder
reaction is frequently employed for the construction of six-membered ring
systems. The high regio- and stereoselectivity typically displayed by this
pericyclic process and the ease of execution have contributed toward its
popularity. Carbon-carbon bond-forming reactions involving N-acyliminium
ions play an extremely important role in the synthesis of nitrogen heterocycles
[3]. A combination of these two powerful synthetic methods would allow
for the rapid, stereocontrolled synthesis of a variety of azapolycyclic
products. This keynote article describes some of our recent work in this
area.
![]() Cycloaddition
of 1,3-Oxazolium-4-oxides
Cycloaddition
of 1,3-Oxazolium-4-oxides
In 1994 we started work in our laboratory to synthesize ring-fused poly-heterocycles based on a sequential cycloaddition-N-acyliminium ion cyclization process [4]. These two types of reaction provide an opportunity for linking two disparate ring forming reactions in a novel sequential manner. We believed that such a protocol would provide one-pot access to target molecules possessing a high degree of complexity which would otherwise require technically demanding multi-step syntheses. Our early studies showed that 1,3-oxazolium-4-oxides (isomünchnones) 2 can be generated by the rhodium(II)-catalyzed cyclization of a suitable diazo imide 1 (Scheme 1) [5]. This type of mesoionic ylide corresponds to the cyclic equivalent of a carbonyl ylide and was found to readily undergo [4+2]-cycloaddition with suitable dipolarophiles [5].
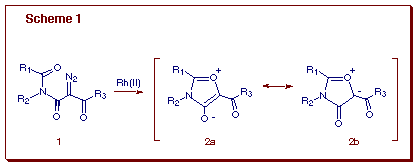
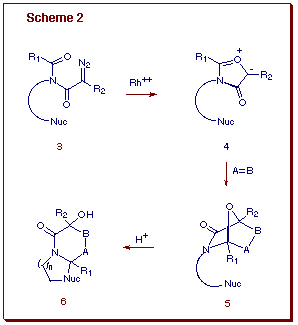
Formation of the isomünchnone ring proceeds by initial generation of a rhodium carbenoid species, followed by an intramolecular cyclization onto the neighboring carbonyl oxygen to form the mesoionic ylide 2. The resultant isomünchnone may be trapped with electron rich or electron deficient dipolarophiles to give the cycloadducts in high yield. These uniquely functionalized cycloadducts (i.e., 5) contain a masked N-acyliminium ion which is generated by its treatment with a Lewis acid [6]. By incorporating an internal nucleophile on the tether, annulation of the original cycloadduct 5 allows for the construction of a more complex nitrogen heterocyclic system, particularly B-ring homologues of the erythrinane family of alkaloids. Starting from simple acyclic diazo imides 3, we established a domino carbenoid cyclization-[4+2]-cycloaddition-cationic ¼-cyclization protocol as a method for the construction of complex nitrogen polyheterocycles of type 6 (Scheme 2). This sequence represents the first example where a [4+2]-cycloaddition and N-acyliminium ion cyclization are coupled in a one-pot sequence. The novelty of the process lies in the method of N-acyliminium ion generation, which to our knowledge, is unprecedented. N-Acyliminium ions are traditionally generated from the N-acylation of imines, N-protonation and oxidation of amides, electrophilic additions to enamides, and the heterolysis of amides bearing a leaving group adjacent to nitrogen [7]. These reactive intermediates readily react with a wide assortment of nucleophiles to effect an overall alpha-amido alkylation.
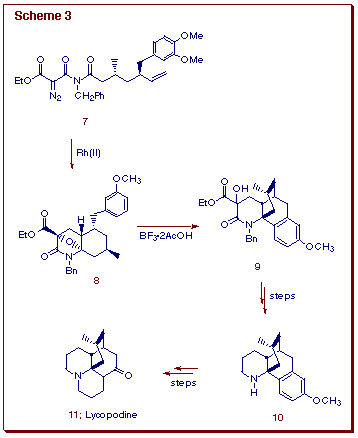
An early application of the domino cascade process toward the construction
of alkaloids involved the synthesis of (±)-lycopodine (11)
(Scheme 3) [8]. The isomünchnone cycloadduct 8 was
formed from the Rh(II)-catalyzed reaction of diazo imide 7 and was
found to be the precursor of the key Stork intermediate 10 (via
9) [9] which was ultimately converted into (±)-lycopodine
11 [9].
![]() Application
of the Domino Cyclization-Cycloaddition Sequence to the Pentacyclic Skeleton
of the Aspidosperma Ring System
Application
of the Domino Cyclization-Cycloaddition Sequence to the Pentacyclic Skeleton
of the Aspidosperma Ring System
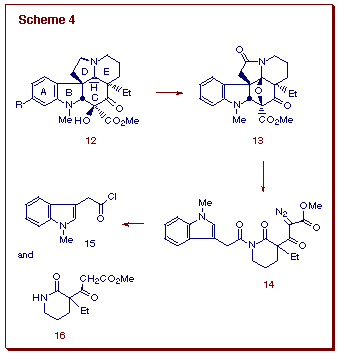
As an extension of these studies, we developed a fundamentally
new approach to the construction of the pentacyclic skeleton of the aspidosperma
ring system which involves a related domino cascade sequence [10]. This
strategy was successfully applied to the synthesis of desacetoxy-4-oxo-6,7-dihydrovindorosine
(12). The approach used is outlined in Scheme 4 and is centered
on the construction of the key oxabicyclic intermediate 13. We reasoned
that 12 should be accessible by reduction of the N-acyliminium
ion derived from 13, which, by analogy with our previous work, should
be available by the tandem rhodium(II)-catalyzed cyclization cycloaddition
of alpha-diazoimide 14. Cycloaddition of the initially formed
dipole across the pendant indole ¼-system [11] would be expected
to result in the simultaneous generation of the CD-rings of the aspidosperma
skeleton [12]. The stereospecific nature of the internal cycloaddition
reaction should results in the correct relative stereochemistry of the
4 chiral centers about the C-ring.
![]() The
2-Aminofuran Diels-Alder Strategy
The
2-Aminofuran Diels-Alder Strategy
During the course of our work in this area, it occurred to us
that we could also utilize a series of 2-amino substituted furans for the
critical [4+2]-cycloaddition step rather than the highly reactive 1,3-dipole.
Our long-range goal involved using 2-amino-substituted furans such as 17
that contain both a suitable leaving group (LG) and an olefinic tether
to allow for an intramolecular Diels-Alder reaction (Scheme 5). The resultant
cycloadduct was expected to undergo ring opening to generate a vinylogous
C-acyliminium ion of type 19. Our intention was to use this sequence
of reactions for a rapid entry into the erythrinane family of alkaloids.
With this goal in mind, some model studies were undertaken to determine
the facility with which 2-aminofurans would undergo internal Diels-Alder
cycloadditions.
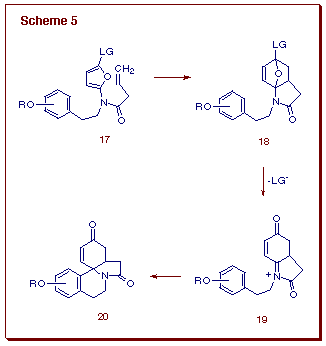
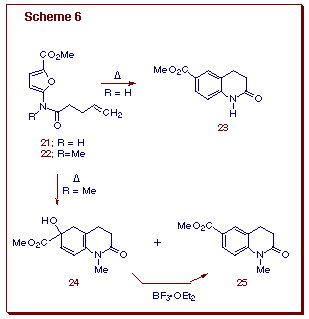
The intramolecular Diels-Alder reaction of furans, often designated as IMDAF, helps to overcome the sluggishness of this heteroaromatic ring system toward [4+2]-cycloaddition [13]. Not only do IMDAF reactions allow for the preparation of complex oxygenated polycyclic compounds, they often proceed at lower temperatures than their intermolecular counterparts. Even more significantly, unactivated ¼-bonds are often suitable dienophiles for the internal cycloaddition. In an effort to investigate the scope of these reactions, a number of new furan substrates were prepared in our laboratory and tested for the cycloaddition-cyclization cascade. Tethered amidofurans 21 and 22 were easily synthesized. The thermal reaction of 21 at 200oC for 24 h afforded tetrahydroquinolinone 23 in 66% yield. Likewise, heating a sample of the N-methylated analog 22 at 160oC furnished a 6:1-mixture of cyclohexadienol 24 (77%) and tetrahydroquinoline 25 (13%), the former being easily converted to 25 by treatment with BF3OEt2. In both cases, the initial cycloadducts were not isolated, as they readily underwent ring opening, assisted by the lone pair of electrons on the adjacent nitrogen (Scheme 6).
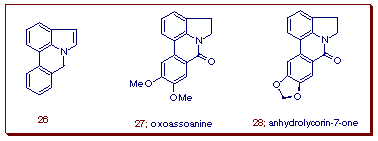
Having established the suitability of 2-amido furans to generate dihydroindoles, we turned our attention to the application of the method toward the synthesis of oxoassoanine [14] (27) and anhydrolycorin-7-one (28) [15]. These compounds are members of the pyrrolophenanthridine class of alkaloids which have been isolated from various species of amaryllidaceae [16]. The 1H-pyrrolo[3,2,1-de]phenanthridine ring system (26) constitutes the core structural framework of the pyrrolophenanthridine alkaloids. Although a number of synthetic routes are available for this ring system, many of these suffer from low yields and lack generality [17].
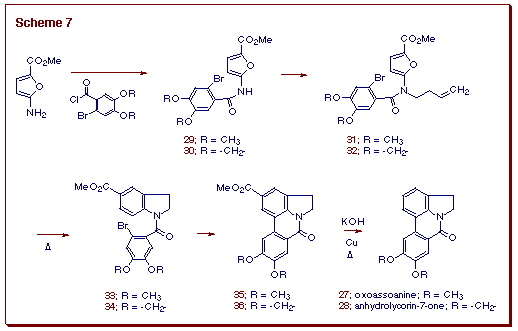
A short synthesis of 27 and 28 was carried out as depicted
in Scheme 7. This approach is centered on the construction of the key dihydroindoles
33 and 34 which are formed by a IMDAF cycloaddition
followed by subsequent nitrogen atom lone pair assisted ring opening of
the initially formed oxabridged cycloadducts. After some experimentation,
it was found that using bis(tributyltin) under photochemical conditions
afforded the aryl-coupled products 35 and 36 in high yield
from the corresponding dihydroindoles 33 and 34. Both compounds
were converted to the natural products by a saponification-decarboxylation
protocol.
![]() The
Domino Pummerer Diels-Alder Sequence
The
Domino Pummerer Diels-Alder Sequence
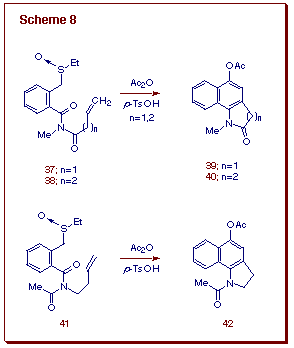
Much of the chemistry utilized in the preceding section relied on our ability to synthesize the requisite 2-aminofurans. One limitation of the method is that sometimes the 2-aminofuran system is not easily accessible. In the context of our studies dealing with domino cycloaddition-Mannich cyclizations, we discovered that the Pummerer reaction can be effectively utilized to prepare the required furans. We focused our attention on an intramolecular variation of the domino amido-Pummerer-Diels-Alder reaction sequence. The one-pot intramolecular cascade process occurred smoothly when the olefin tether was activated by an ester or when a carbonyl group was located adjacent to the nitrogen atom of the 2-amino substituted isobenzofuran (Scheme 8) [18]. The intramolecular cycloaddition behavior of the incipient isobenzofurans in response to the presence of a C=O group is striking. Five and six ring-membered precursors 37 and 38 delivered cyclized products bearing a carbonyl within the newly formed rings in good to excellent yields.
![]() Triple
Cascade Sequence for Construction of the Erythrinane Alkaloid Skeleton
Triple
Cascade Sequence for Construction of the Erythrinane Alkaloid Skeleton
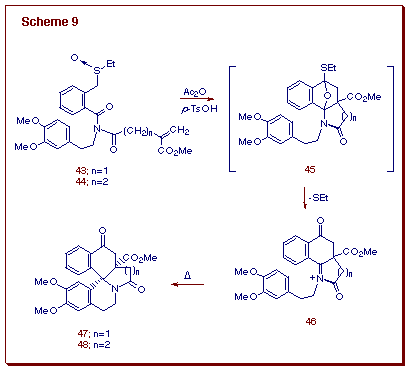
Having established the facility with which N-acyliminium ions can be formed from the Pummerer reaction of o-amido substituted sulfoxides, we next focused our attention on the final cyclization step of the proposed cascade process (i.e., 19 -> 20 in Scheme 5) [19]. In order to avoid the deprotonation (aromatization) step, we prepared sulfoxides 43 and 44, each possessing a carbomethoxy group attached to the olefin tether. This substituent was selected not only to prevent deprotonation, but also because the presence of an electron withdrawing group on the double bond enhances [4+2]-cycloaddition based on FMO considerations. N-Acyliminium ion 46 derived from the internal cycloadduct 45 underwent stereoselective spirocyclization to furnish the cis-3,4-benzoerythrinane 47 or homoerythrinane derivative 48 in good yield (Scheme 9). The overall triple cascade sequence represents an efficient one-pot approach towards the erythrinane alkaloid skeleton in which the spirocyclic ABC skeleton is assembled in a single operation.
At this point, we decided to undertake a synthesis of (±)-erysotraamidine (58) in order to further test the viability of the triple cascade process as an entry into the erythrinane skeleton. The requisite starting imidosulfoxide 49, possessing both a dienophilic and diactivated aromatic ¼-tether, was efficiently synthesized from known starting materials. Subjection of 49 to the Pummerer conditions gave compound 55 as a single diastereomer in 83% yield. The cis A/B ring fusion present in 55 was unequivocally established by an X-ray crystallographic analysis and is identical to the stereochemical relationship found in the naturally occurring Erythrina alkaloids. The conversion of 49 into 55 is believed to follow the pathway outlined below (Scheme 10). The initially formed alpha-thiocarbocation intermediate generated from the Pummerer reaction of 49 is intercepted by the adjacent imido carbonyl to producethe alpha-amido substituted furan 50.
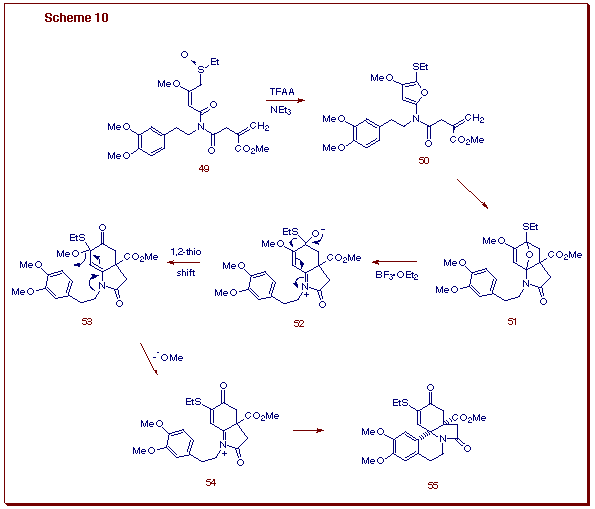
This transient intermediate undergoes a subsequent intramolecular Diels-Alder
cycloaddition across the tethered ¼-bond to furnish cycloadduct
51. Nitrogen-assisted ring opening of the oxabicyclic bridge results
in the formation of zwitterionic intermediate 52 which undergoes
a 1,2-thioethyl shift followed by methoxide ion ejection. Cyclization of
the diactivated aromatic tether onto N-acyliminium ion 54 ultimately
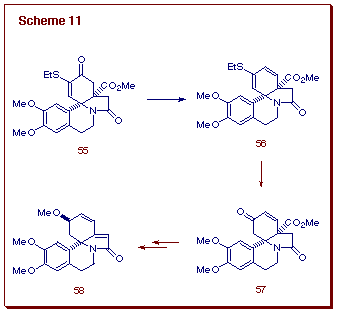
provides the tetracyclic amide 55. With a supply of 55 in
hand, this enone was converted into the corresponding vinyl triflate which,
in turn, was subjected to a palladium catalyzed formate reduction to give
56. The resulting thio-substituted diene was subsequently transformed
into ketone 57 via a titanium mediated hydrolysis. The present
sequence constitutes a formal synthesis of (±)-erysotramidine (58)
based on the successful conversion of 57 into 58 by Tsuda
and coworkers (Scheme 11) [20].
![]() Concluding
Remarks
Concluding
Remarks
Over the past several years we have shown that many structurally diverse
heterocyclic compounds can be readily accessed via the domino
cycloaddition/N-acyliminium ion cyclization cascade. The key step in
this process involves the generation of a reactive N-acyliminium
ion by fragmentation of an amino substituted [4+2]-cycloadduct. This triple
cascade is applicable toward the preparation of a broad range of alkaloids.
It is a reasonable expectation that future years will see a continued evolution
of this unique domino cascade toward other synthetic targets.
![]() Acknowledgment
Acknowledgment
I thank my enthusiastic and gifted coworkers whose names
appear in the appropriate references and the National Institute of Health
for financial support.
![]() References
References
1. Denmark, S. E.; Thorarensen, A. Chem. Rev.
1996, 96, 137.
2. Winkler, J. D. Chem. Rev. 1996, 96, 167. Roush, W. R. In Comprehensive Organic Synthesis; Trost, B. M.; Pergamon Press: New York, 1991; Vol. 5, p 513. Padwa, A., Ed. 1,3-Dipolar Cycloaddition Chemistry; Wiley-Interscience: New York, 1984; Vols. I and II.
3. Hiemstra, H.; Speckamp, W. N. In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon: Oxford, 1991; Vol. 2, pp 1047-1082.
4. Marino, J. P., Jr.; Osterhout, M. H.; Price, A. T.; Semones, M. A.; Padwa, A. J. Org. Chem. 1994, 59, 5518.
5. Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223. Padwa, A.; Marino, J. P. Jr.; Osterhout, M. H. J. Org. Chem. 1995, 60, 2704. Padwa, A.; Hertzog, D. L.; Nadler, W. R.; Osterhout, M. H.; Price, A. T. J. Org. Chem. 1994, 59, 1418. Hertzog, D. L.; Austin, D. J.; Nadler, W. R.; Padwa, A. Tetrahedron Lett. 1992, 33, 4731. Osterhout, M. H.; Nadler, W. R.; Padwa, A. Synthesis 1994, 123.
6. Padwa, A.; Brodney, M. A.; Marino, J. P. Jr.; Osterhout, M. H.; Price, A. T. J. Org. Chem. 1997, 62, 67.
7. Speckamp, W. N.; Hiemstra, H. Tetrahedron 1985, 41, 4367.
8. Padwa, A.; Brodney, M. A.; Marino, J. P., Jr.; Sheehan, S. M. J. Org. Chem. 1997, 62, 78.
9. Stork, G.; Kretchmer, R. A.; Schlessinger, R. H. J. Am. Chem. Soc. 1968, 90, 1647. Stork, G. Pure Appl. Chem. 1968, 17, 383.
10. Padwa, A.; Price, A. T. J. Org. Chem. 1995, 60, 6258. Padwa, A.; Price, A. T. J. Org. Chem. 1998, 63, 556.
11. Padwa, A.; Hertzog, D. L.; Nadler, W. R. J. Org. Chem. 1994, 59, 7072.
12. Cordell, G. A. In The Alkaloids; Manske, R. H. F., Rodrigo, R. G. A., Eds.; Academic Press: New York, 1979, Vol. 17, pp 199-384. Saxton, J. E. Nat. Prod. Rep. 1993, 10, 349; ibid 1994, 11, 493.
13. Kappe, C. O.; Murphree, S. S.; Padwa, A. Tetrahedron, 1997, 53, 14179. Woo, S.; Keay, B. Synthesis 1996, 669. Dean, F. M. Adv. Heterocycl. Chem. 1981, 30, 168.
14. Llabres, J. M.; Viladomat, F.; Bastida, J.; Codina, C.; Rubiralta, M. Phytochemistry 1986, 25, 2637.
15. Ghosal, S.; Rao, P. H.; Jaiswal, D. K.; Kumar, Y.; Frahm, A. W. Phytochemistry 1985, 24, 1825. Perez, D.; Bures, G.; Guitian, E.; Castedo, L. J. Org. Chem. 1996, 61, 1650.
16. Hill, R. K.; In The Alkaloids; Manske, R. H. F., Ed.; Academic Press: New York, 1967; Vol. 9, p 483.
17. Hutchings, R. H.; Meyers, A. I. J. Org. Chem. 1996, 61, 1004 and references cited therein.
18. Padwa, A.; Kappe, C. O.; Cochran, J. E.; Snyder, J. P. J. Org. Chem. 1997, 62, 2786.
19. Padwa, A.; Kappe, C. O.; Reger, T. S. J. Org. Chem. 1996, 61, 4888. Padwa, A.; Hennig, R.; Kappe, C. O.; Reger, T. S. J. Org. Chem. 1998, 63, 1144.
20. Tsuda, Y.; Hosoi, S.; Nakai, A.; Sakai, Y.; Abe, T.; Ishi, Y.; Kiuchi, F.; Sano, T. Chem. Pharm. Bull. 1991, 39, 1365.