| [Related articles/posters: 046 089 023 ] |
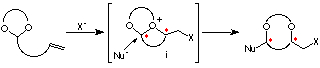
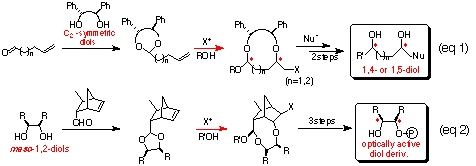
In this conference, we present two types of embodiments, asymmetric synthesis of optically active 1,4- and 1,5-diols (eq 1, Chapter 1),1 and desymmetrization of meso-1,2-diols (eq 2, Chapter 2).2
Chapter 1: Asymmetric Synthesis of Optically Active 1,4- and 1,5-Diols
Introduction
Nucleophilic addition to a carbocation or an oxonium ion, formed by a C-O bond fission of an acetal, has been recognized as a useful synthetic tool in organic chemistry, and many variants have been developed so far. In these reactions, a Lewis acid is usually used as an initiator to accelerate the cleavage of the acetal.3 Cationic species formed by electrophilic addition of a halonium ion to a double bond, have also been utilized to cleave an acetal for instance, Fraser-Reid et al. had recently reported that a remote double bond can cleave an acetal under neutral, oxidative conditions through an intramolecular haloetherification reaction.4 On the other hand, asymmetric synthesis using chiral acetals from C2-symmetric diols has been intensively studied as an attractive approach to optical active compounds.5 This transformation can generally be classified into two types: 1) asymmetric reactions involving cleavage of the C-O bond of the acetal, which functions as a chiral synthetic equivalent to a carbonyl group, and 2) asymmetric induction on a neighboring prochiral center. However, no report involving a combination of these two types of reactions in the same system has been published so far. In addition, in the type-1 reactions, generally only Lewis acids have been used to cleave the acetal group.
It occurred to us that the intramolecular haloetherification reaction of the ene acetal 1, prepared from a C2-symmetric diol, in the presence of an alcohol could proceed through a cascade of cationic intermediates ii and iii, to give a cyclic acetal with two newly formed asymmetric centers. This novel methodology involving two types of reaction using C2-symmetric acetals is unprecedent (Scheme 1).6
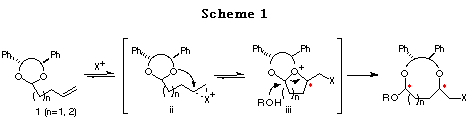
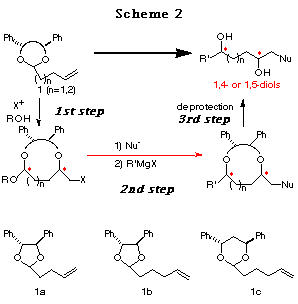
In order to develop this chemistry, we examined the transformation of several ene acetals. Among them, the ene acetal 1a (Table 1) derived from chiral hydrobenzoin (n=1) and the ene acetal 1c (Table 2) derived from chiral 1,3-diphenyl-1,3-propanediol (n=2) were found to be the best substrates for affording cyclic mixed acetals with high diastereoselectvity (Scheme 2, 1st step). After substituting the iodide group by a nucleophile, the alkoxy group of the resulting mixed acetal was converted to an alkyl group using a Grignard reagent (Scheme 2, 2nd step). Finally, deprotection of the diphenylethylene or diphenylpropylene unit afforded chiral 1,4- or 1,5-diols, respectively (Scheme 2, 3rd step). We present in this chapter the full details of our results7 and the formal synthesis of Solenopsin A,8 which is isolated from the venom secreted by the fire ant Solenopsis invicta, and the synthesis of a civet constituent.9
Results and Discussion
1st step:
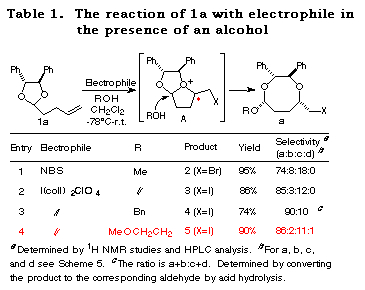
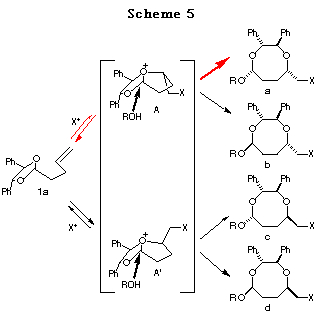
Haloetherification reaction. Initially, the haloetherification reaction of 1a which has a chiral hydrobenzoin10 as an auxiliary was studied. Electrophiles such as NBS and I(coll)2ClO411 gave the cyclic mixed acetals 2-5 in the presence of ROH. The reaction using I2 gave the product of addition of I2 to the double bond. As shown in Table 1, I(coll)2ClO4 proved to be a better choice than NBS in terms of diastereoselectivity (entry 1 vs 2). Several alcohols were examined in this reaction and the stereoselectivities are similar (entries 2-4). The structures of the major products (a) were determined by 1H NMR study and the conversion to known compounds12 and shown in Table 1 (for the structures of b, c and d, see Scheme 5).13
Furthermore, the reaction of 1b prepared from chiral hydrobenzoin and 5-hexenal was studied. In this case, only normal haloetherification reaction products 6 were obtained with no diastereoselectivity (Scheme 3).
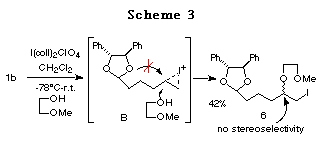
The difference in the reactivity between 1a and 1b can be rationalized as follows: 1) The longer distance between the acetal oxygen atom and the cation formed on the double bond in 1b prevents the formation of a bicyclic cationic intermediate like A in 1a,14 and 2) the 5,6-membered bicyclic cationic intermediate B' is more unstable than the 5,5-membered one A. Hence, we prepared the ene acetal 1c derived from chiral 1,3-diphenyl-1,3-propanediol,15 with the hope that the 6,6-membered bicyclic intermediate C could be more stable and more easily formed (Scheme 4).16
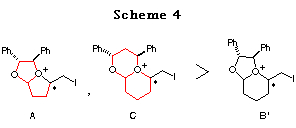
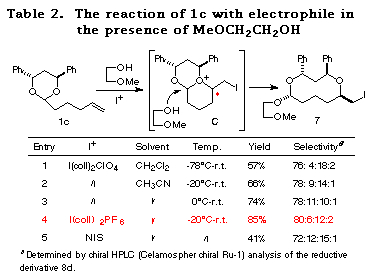
As expected, although a higher temperature (-20ÁC) was necessary compared to 1a (-78ÁC) and the selectivity was slightly reduced, the reaction of 1c proceeded successfully to give the cyclic acetal 7 in good yields (Table 2). In this case, CH3CN is the solvent of choice (entry 1 vs. 2) and bis(sym-collidine)iodine hexafluorophosphate I(coll)2 PF617 showed the best stereoselectivity and reactivity (entry 4). The structure of major product 7 was determined by conversion to the known compounds 11 and 15 and by postulating that the reaction proceeded through an SN2-type attack of the alcohol to the bicyclic oxonium ion C as observed in the reaction of 1a (see the section of reaction mechanism).
Reaction mechanism. The plausible reaction mechanism in the case of
1a is shown in Scheme 5. Initial addition of the halonium ion to
the double bond followed by attack of one of the acetal oxygen atoms forms
the bicyclic oxonium ion intermediates A or A'. Subsequent
SN2-type attack of an alcohol to A or A' would give the 8-membered
acetal (a>b, c>d). In view of the fact
that this reaction gives rise exclusively to an 8-membered acetal a
as a major product, it could be postulated that intermediate A should
be more favorable than A' (Scheme 5, a+b>c+d).
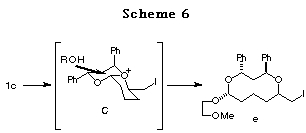
In the case of 1c, the reaction might proceed in a similar way through
the most stable intermediate C among the possible bicyclic intermediates
(Scheme 6).18
2nd step:
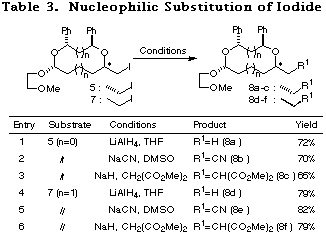
Substitution of iodide and alkoxy group. Substitution of the iodide in compounds 5 and 7, involving four stereoisomers respectively, with several nucleophiles proceeded successfully using known procedures. The results are summarized in Table 3. At this stage, 8a-f were obtained in a pure state and the yields are shown in Table 3 for pure 8a-f.
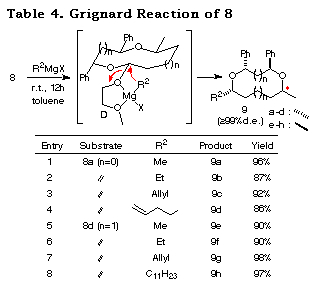
We next studied the nucleophilic replacement of the alkoxy group of 8a and 8d with carbon nucleophiles. Grignard reagents19 reacted at the acetal carbon and afforded the corresponding substituted products 9 with complete retention of stereochemistry (Table 4).20 A long chain carbon nucleophile can be introduced without any difficulty (entry 8). In these reactions, the methoxyethoxy group is essential since the derivatives with a methoxy group or benzyloxy group derived from 3 or 4 by LiAlH4 reduction showed disappointing results (with MeMgI; 43% for the methoxy derivative and 0% for the benzyloxy derivative). The reason for the good yields and complete retention of stereochemistry can be attributed to the formation of complex D. Under Lewis acidic conditions (TiCl4/allyltrimethylsilane/CH2Cl2/-78ÁC) the reaction gave complex mixtures.
3nd step:
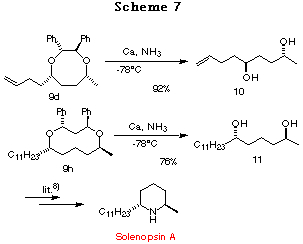
Deprotection. Having achieved highly stereoselective construction of compounds 9a-h, we then examined the removal of the chiral auxiliary unit using catalytic hydrogenolysis or Birch reduction. Both processes proceeded smoothly and afforded the corresponding optically active 1,4- and 1,5-diols 10, 11 in good yields, respectively. Since the 1,5-diol 11 had already been converted to solenopsin A, a constituent of the venom of the fire ant Solenopsis invicta, by G. Solladi et al.,8 a formal synthesis of Solenopsin A was achieved (Scheme 7).
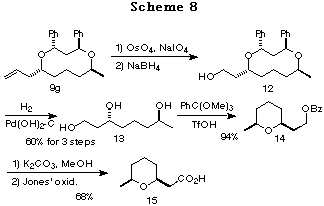
Synthesis of a civet constituent. Synthesis of (+)-(S,S)-(cis-6-methyltetrahydropyran-2-yl)acetic acid 15, a civet constituent that was isolated from the perfume material civet,21 a glandular secretion of the civet cat Viverra civetta was also achieved. Oxidative cleavage of the olefin of 9g followed by NaBH4 reduction gave the alcohol 12. Catalytic hydrogenolysis of 12 afforded the triol 13, which was combined with PhC(OMe)3 to give the cyclic ether 14.22 Alkaline hydrolysis of 14 followed by Jones' oxidation gave a civet constituent, the cyclic ether 15.
Conclusions
A new asymmetric synthesis of chiral 1,4- and 1,5-diols has been developed based on remote asymmetric induction as a key step. In the 1st step, the acetal acts initially as a nucleophile and then as an electrophile in a one-pot operation and the two remote stereogenic centers are built up simultaneously. It is noteworthy that the formation of a bicyclic cationic intermediate makes the reaction stereoselective. In the 2nd step, the conversion of an alkoxy group to alkyl groups has been achieved in high yields with complete retention of stereochemistry. The use of a methoxyethoxy functionality is noteworthy. In the 3rd step, the usual deprotection procedure worked well to give optically active 1,4- and 1,5-diols. These results suggest that the diphenylethlene or diphenylpropylene unit is an efficient and useful protecting group for diols.
Chapter 2: Desymmetrization of meso-1,2-Diols
Introduction
Desymmetrization of the °Ü-symmetric diols (including meso-diols) is an important area in organic synthesis. Many methodologies have been developed so far. They can be widely classified into 2 types, one based on the use of enzymes23 and the other based on conventional chemical methods.24 Although both of them have proved to be very useful in desymmetrization of prochiral diols, no successful report on highly enantioselective deymmetrization of acyclic meso-1,2-diols has appeared, to the best of our knowledge. In this chapter we present here a conceptionally novel and highly enantioselective desymmetrization of meso-1,2-diols, which is very powerful not only for cyclic diols but also for acyclic ones.
Results and Discussion
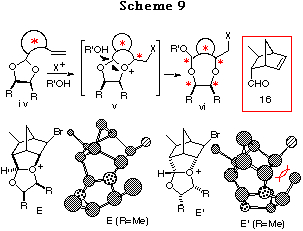
In Chapter 1, a new asymmetric synthesis of optically active 1,4- and 1,5-diols has been highlighted, wherein intramolecular haloetherification of chiral ene acetals, prepared from C2-symmetric optically active diols and ene aldehydes, forms the crucial step. The reaction of an ene acetal proceeds via an oxonium ion intermediate.1 This finding suggested that if a large energy difference existed among the possible intermediates v formed from the ene acetal iv derived from the proper chiral non-racemic ene aldehyde and a symmetric meso-diol, the reaction may proceed through the most stable intermediate resulting in the discrimination of the two oxygen atoms of the meso-diol. We selected (1R,2R,3S,4S)-3-methyl-5-norbornene-2-carboxaldehyde 16 as a chiral ene aldehyde for the following four reasons: (1) 16 is easily prepared by asymmetric Diels-Alder reaction,25 (2) acetalization would proceed stereoselectively to give the cis-isomer,26 (3) a newly produced chiral center (* center in v) would be formed stereospecifically in the haloetherification because the double bond is fixed in the ring, and the most importantly (4) sterically rigid forms of the oxonium ion intermediates would be expected to cause a large energy difference. Support for (4) arises from the consideration of the discriminating process for the two oxygen atoms of the acetal. As will be described later, because acetalization of 16 with meso-diols proceeded stereoselectively to give cis-ene acetals, two possible cis-intermediates,27 E and E', are depicted. As shown from the conformations of the intermediates E (R=Me) and E' (R=Me) obtained by 'energy minimization' using the simple CS Chem 3D Pro Program (MM2 force field), a large steric repulsion not only between the substituents and the bicyclo[2.2.1]heptane skeleton but also between the 1,3-dioxolane skeleton and the bicyclo[2.2.1]heptane skeleton is observed in endo isomer E' whereas such a large steric repulsion is not observed in exo isomer E. This must suggest a large energy difference, in other words a large stability difference, between the two cis-intermediates E and E', realizing an extremely high discrimination of the two oxygen atoms of the acetal regardless of a cyclic or acycic system.
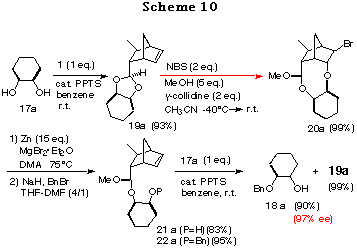
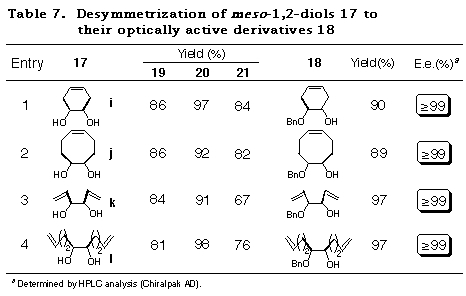
To realize the above concept, meso-cyclohexane-1,2-diol 17a was examined as a substrate (Scheme 10). The cis-ene acetal 19a, readily synthesized as a single isomer in 93% yield from 16 and 17a, was subjected to an intramolecular haloetherification to give the mixed acetal 20a via an oxonium ion intermediate (refer to intermediate E in Scheme 9). As expected, it was confirmed to be a single isomer by 1H NMR data. This clearly showed that the discriminating process proceeded in a quite highly stereoselective manner. Dehaloetherification of 20a using Zn and MgBr2´Et2O afforded the acetal 21a in good yield. Protection of the hydroxy group of 21a as a benzyl ether followed by transacetalization with one equivalent of meso-diol 17a furnished the monoprotected diol 18a in good yield. At the same time, 19a was regenerated quantitatively. The optical purity of 18a was determined to be 97% ee by HPLC analysis with a Daicel Chiralpak AD. The absolute stereochemistry of 18a was determined by comparison of its specific rotation [+16.6Á(c 1.03, CHCl3)] with the reported value [+16.5Á(c 1.10, CHCl3)],28 which was in good agreement with the structure expected from the consideration of the stabilities of the intermediates in Scheme 9.
The success of the desymmetrization of 17a affording optically active 18a in high enantiomeric excess and simultaneous regeneration of 19a offers an efficient reaction cycle for desymmetrization of meso-1,2-diols 17 to their optically active derivatives 18 as depicted in Scheme 11.
The applicability of this method to various kinds of meso-1,2-diols, cyclic and acyclic ones was then investigated. The results of the desymmetrization of 17a was considered for comparison. Attempts to acetalize 16 with meso-1,2-diols 17a-h were successful and gave only single diastereomers, namely cis-isomers 19a-h, almost quantitatively in each case (Table 5). The stereochemistry of 19a-h were determined by their NOE measurements.
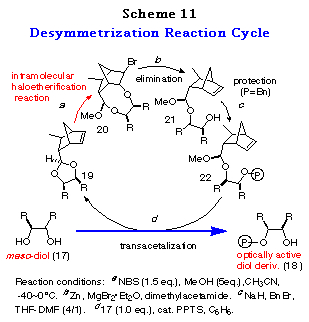
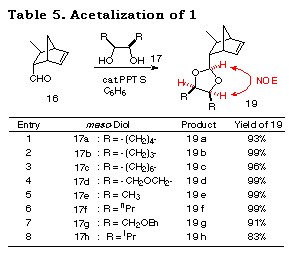
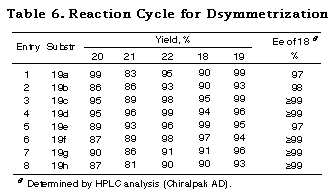
The results subjecting the ene acetals 19 to the desymmetrization reaction cycle (Scheme 3) are shown in Table 2. Intramolecular haloetherification of 19 to gave the mixed acetals 20 as a single isomer determined by 1H NMR in each case. Dehaloetherification, benzylation, and transacetalization afforded the optically active 18 and 19. All the reaction steps proceeded in high yields. The optical purity of 18 was extremely high (>97% ee) irrespective of ring size and the presence of an oxygen atom in the cyclic systems (entries 1-4) and of the nature of substituent in acyclic ones (entries 5-9). The absolute stereochemistry of the compounds was deduced from the mechanistic consideration in Scheme 9 and the following ones; that of cyclic monoprotected diols 18b-d was determined by assuming the same sense of stereoselection as observed for 18a and that of acyclic ones 18e-i was determined from the consideration of X-ray crystallographic structure of the analog of 20e.29
The other feature of the method is easy choice of a proper protecting group. For example, in the case of the diol 17g having a benzyl group, the silyl, acyl, or p-methoxybenzyl (MPB) group can also be used as protecting groups. Thus 21g was converted to the silyl, acyl and p-methoxybenzyl compounds 24a,b,c, whose ee values were estimated to be >99%,30 in good yields without any problem (Scheme 12).
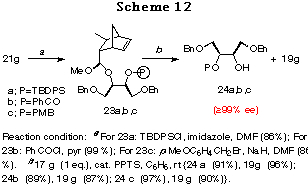
Since the crucial step of this desymmetrization cycle was an intramolecular haloetherification reaction, only saturated diols were examined in Table 6. It was envisaged that if the methodology could be extended to the diols having olefins in the same molecule, namely unsaturated ones (Figure 1), the method would find application for the construction of more useful chiral building blocks.
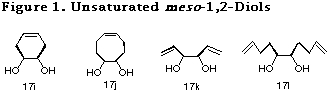
Although, similar haloetherification reaction of the ene acetals derived from unsaturated diols (i.e. presence of double bond in 19) was anticipated to bring about a competition between the intra- and intermolecular reaction pathway, the following examination showed us a remarkable kinetic control preferable for the intramolecular reaction over the intermolecular one, providing various unsaturated optically active diol derivatives by the same desymmetrization reaction cycle.
To begin with, we investigated the reaction of 4-cyclohexene-1,2-diol 17i in order to examine the possibility of the use of unsaturated diols in our desymmetrization method. Haloetherification reaction of 19i31 derived from 17i in the presence of MeOH, to our surprise, proceeded only in an intramolecular fashion to give the 8-membered acetal 20i in a quite high yield. Interestingly, product arising out of the reaction of cyclohexene olefin was not at all observed. The degree of the desymmetrization of two oxygen atoms of acetal 19i was determined to be 100% by NMR experiment. Its stereochemistry was established by converting it to the known compound via hydrogenation. This extremely high stereoselectivity in haloetherification reaction is probably due to a large stability difference between the two cis-intermediate, since endo-isomer has a large steric repulsion not only between the substituents and the bicyclo[2.2.1]heptane skeleton but also between the 1,3-dioxolane skeleton and the bicyclo[2.2.1]heptane skeleton as discussed in our earlier paper.2
In order to show the versatility of the remarkable kinetic control of this haloetherification reaction, we then examined the reactions of the acetals 19j31 and 19k31 derived from 5-cyclooctene-1,2-diol 17j and 1,5-hexadiene-3,4-diol 17k. In both the cases, high kinetic control was observed and the desired products 20j and 20k were obtained in extremely high yields through the intramolecular reaction. High selectivity in these haloetherification reaction could be due to the preference of the intramolecular reaction over that of the intermolecular one, in addition to the high reactivity of norbornene olefin to form the bromonium ion because of its non-hyperconjugated character.
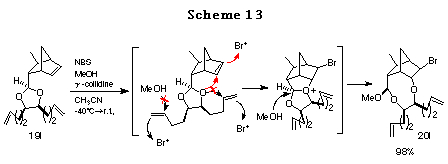
To ascertain the high reactivity of norbornene olefin, we chose the ene acetal derived from 1,9-decadiene-5,6-diol 19l.31 In this case, since both the norbornene olefin and the diol olefin are equidistant from the acetal oxygen and in addition, both the olefins are expected to form a 5-membered transition state with the acetal oxygen, it is of great interest to examine their relative reactivities. As expected, the reaction proceeded exclusively with the norbornene olefin to give the desired product 19l in high yield. This chemoselective haloetherification reaction is quite noteworthy from synthetic point of view (Scheme 13).
Having achieved the desymmetrization of unsaturated meso-diols 17 in quite high diastereoselectivity by remarkable kinetic control of haloetherification reaction of ene acetals 19, we then focussed our attention on the conversion of the mixed acetals 20 to the optically active monoprotected diol derivative 18 as per the procedure reported previously by us32 (refer to Scheme 11). Table 7 shows the results of overall conversion of meso-diols 17 to their optically active derivatives 18 including the yields of acetalization and intramolecular haloetherification. It should be pointed out that MgBr2´Et2O, which has been reported by us as an additive during the dehaloetherification reaction, was found to be inffective in the present case. On the other hand, ZnCl2 proved to be an efficient additive for the dehaloetherification of the mixed acetal 20. A point worth mentioning is that both the acid labile allylic ether moieties present in 20l remained infact under the reaction conditions. The subsequent two step sequence, viz., protection of the hydroxy group as benzyl ether and transacetalization with the starting meso-diols 17, afforded the desired optically pure unsaturated meso-1,2-diol derivatives 18, with the regeneration of the ene acetals 19 (84~90%), whose optical purities were ascertained by HPLC analysis.
Conclusions
In summary, we have developed a new desymmetrization method for meso-1,2-diols. Characteristic points of the method are (i) desymmetrization with an extremely highly enantiomeric excess, (ii) wide applicability not only to cyclic1,2-diols but also to acyclic ones for which no successful report has appeared yet, (iii) ready protection with proper group, and (iv) high efficiency through the desymmetrization reaction cycle. In addition to that, we have developed an efficient desymmetrization reaction of unsaturated meso-1,2- diols with a high degree of kinetic control. Chemoselective haloetherification of norbornene olefin in presence of other double bonds is a highly remarkable finding, as the unreacted olefin moiety could be exploited for further elaboration, leading to the synthesis of a variety of optically active compounds.
References and Notes
(1) (a) Fujioka, H.; Kitagawa, H.; Matsunaga, N.; Nagatomi, Y.; Kita, Y. Tetrahedron Lett. 1996, 37, 2245. (b) Fujioka, H.; Kitagawa, H.; Nagatomi, Y.; Kita, Y. J. Org. Chem. 1996, 61, 7309.
(2) Fujioka, H.; Nagatomi, Y.; Kitagawa, H.; Kita, Y. J. Am. Chem. Soc. 1997, 119, 12016.
(3) For examples, see: (a) Kotsuki, H. Synlett 1992, 97. (b) Mukaiyama, T.; Murakami, M. Synthesis 1987, 1043. (c) Mukaiyama, T. Org. React. 1982, 28, 203.
(4) For studies of halonium ion mediated neighboring group activation of acetal cleavage, see: (a) Robert, M.; Udodong, U. E.; Roberts, C.; Mootoo, D. R.; Konradsson, P.; Fraser-Reid, B. J. Am. Chem. Soc. 1995, 117, 1554. (b) Roberts, C.; Madsen, R.; Fraser-Reid, B. J. Am. Chem. Soc. 1995, 117, 1546. (c) Madsen, R.; Fraser-Reid, B. J. Org. Chem. 1995, 60, 772. (d) Fraser-Reid, B.; Udodong, U. E.; Wu, Z.; Ottosson, J.; Merritt, J. R.; Rao, C. S.; Roberts, C. Synlett 1992, 927. (e) Zhang, H.; Wilson, P.; Shan, W.; Ruan, Z.; Mootoo, D. R. Tetrahedron Lett. 1995, 36, 649. (f) Wilson, P.; Shan, W. F.; Mootoo, D. R. J. Carbohydrate Chem. 1994, 13, 133. (g) Elvey, S. P.; Mootoo, D. R. J. Am. Chem. Soc. 1992, 60, 9685. (h) Mootoo, D. R.; Date, V. Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1987, 1462. References e-h include the reactions of remote stereocontrol.
(5) For reviews on the asymmetric synthesis using the chiral C2-symmetric acetals, see: (a) Alexakis, A.; Mangeny, P. Tetrahedron: Asymmetry, 1990, 1, 477. (b) Fujioka, H.; Kita, Y. "Studies in Natural Product Chemistry", ed by Atta-ur-Rahman, Elsevier, Amsterdam, 1994; Vol 14, pp 469.
(6) For our representative studies using the chiral C2-symmetric acetals, see: (a) Fujioka, H.; Kitagawa, H.; Yamanaka, T.; Kita, Y. Chem. Pharm. Bull. 1992, 40, 3118. (b) Fujioka, H.; Annoura, H.; Murano, K.; Kita, Y.; Tamura, Y. Chem. Pharm. Bull. 1989, 37, 2047. (c) Tamura, Y. Annoura, H.; Yamamoto, H.; Kondo, H.; Kita, Y. Fujioka, H. Tetrahedron Lett. 1987, 28, 5709. (d) Tamura, Y. Annoura, Fujioka, H. Tetrahedron Lett. 1987, 28, 5681. (e) Tamura, Y.; Ko, T.; Kondo, H.; Annoura, H.; Fuji, M.; Takeuchi, R.; Fujioka, H. Tetrahedron Lett. 1986, 27, 2117. (f) Tamura, Y.; Kondo, H.; Annoura, H.; Takeuchi, R.; Fujioka, H. Tetrahedron Lett. 1986, 27, 81.
(7) The preliminary stereoselective synthesis of optically active 1,4-diols has been recently reported by us: Fujioka, H.; Kitagawa, H.; Matsunaga, N.; Nagatomi, Y.; Kita, Y. Tetrahedron Lett. 1996, 37, 2245.
(8) Solladi, G.; Huser, N. Recl. Trav. Chim. Pays-Bas. 1995, 114, 153 and references therein.
(9) For previous synthesis of the optically active natural product, see: (a) Keinan, E.; Seth, K. K.; Lamed, R. J. Am. Chem. Soc. 1986, 108, 3474. (b) Seebach, D.; Pohmakotr, M. Helv. Chim. Acta 1979, 62, 1096. (c) Lichtenthaler, F. W.; Klingler, F. D.; Jarglis, P. Carbohydr. Res. 1984, 132, C1.
(10) Chiral hydrobenzoin is readily available in both enantiomeric forms by asymmetric synthesis or resolution; for asymmetric synthesis, see, Wang, Z. -M.; Sharpless, K. B. J. Org. Chem. 1994, 59, 8302, and for resolution of dl-hydrobenzoin, see "Optical Resolution Procedures for Chemical Compounds" ed. by P. Mewman, Optical Information Center, Manhattan College, Riverdale, New York 10471, 1984, Vol. 3, pp 353.
(11) Preparation: Lemieux, R. U.; Morgan, A. R. Can. J. Chem. 1965, 43, 2190.
(12) The stereochemistries of the major products a were determined from the NOE experiment on 8a (Table 3) derived from 5a. The stereochemistries of the halomethyl substituent were determined by converting 5 to the (R)-enriched-pentane-1,4-diol {[°Ŕ]D23 -10Á (c 0.46, MeOH) lit. [°Ŕ]D21 -13.4Á (c 1.05, MeOH); Kitahara, T.; Mori, K.; Matsui, M. Tetrahedron Lett. 1979, 32, 3021.} in a three-step sequence; 1) acid hydrolysis, 2) LiAlH4 reduction, and 3) hydrogenolysis.
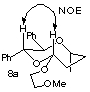
(13) The structures of b, c and d were postulated from consideration of the reaction mechanism and the assignment is tentative.
(14) A similar result was reported by Fraser-Reid et al. They suggested that the formation of the 6-membered oxonium ion was slower than that of the 5-membered one (see ref. 4d).
(15) Optically active 1,3-diphenyl-1,3-propanediol is available in both enantiomeric forms by asymmetric synthesis or microbial resolution; for asymmetric synthesis, see, Wang, Z. -M.; Ito, K.; Harada, T.; Tai, A. Bull. Chem. Soc. Jpn. 1980, 53, 3367, and for microbial resolution, see, Yamamoto, K.; Ando, H.; Chikamatsu, H. J. Chem. Soc., Chem. Commun. 1994, 334.
(16) There is another possible 6,5-membered bicyclic intermediate from reaction of the acetal derived from 4-pentenal and 1,3-diphenyl-1,3-propanediol. We didn't feel it necessary to examine its reaction in order to develop the asymmetric synthesis of 1,4-diols.
(17) Bis(sym-collidine)iodine(I) hexafluorophosphate (I(coll)2PF6) is also used during halolactonization, see, Simonot, B.; Rousseau, G. J. Org. Chem. 1994, 59, 5912.
(18) We performed MO calculations for all of possible 6,6-membered bicyclic oxonium ions by SPARTAN (ver. 3.1.2) using the AM1 Hamiltonian. The results showed that the cis-decaline form intermediate C is the most stable among the others involving trans-decaline form intermediates (The heat of formation of C was 145.71 kcal/mol. The others were 146.80 kcal/mol for another cis-decaline form intermediate and 148.79, 151.16 kcal/mol for trans-ones). These calculational results were consistent with our experimental results. We think that the stability of bicyclic intermediates has a large influence in the reaction of 1c. We also performed MO calculations on 5,5-membered bicyclic intermediates. In these cases, the intermediate A' was more stable than A (The heat of formation of A was 159.30 kcal/mol, and of A' was 157.18 kcal/mol). This calculational result did not fit with our experimental results. It is probable that the repulsion between the halomethyl substituent and anion species, which should exist on the convex side of the cation has a large influence in the reaction of 1a and makes the formation of A' undesirable.
(19) For the reaction of a Grignard reagent and an acetal, see: Yuan, T. -M.; Yeh, S. -M.; Hsieh, Y. -T.; Luh, T. -Y. J. Org. Chem. 1994, 59, 8192 and references therein.
(20) The stereochemistry of compounds 9a-h were determined by X-ray crystallography of the p-nitrobenzoate derivative derived from 9d by hydroboration-oxidation followed by p-nitrobenzoylation. The author has deposited atomic coordinates for this structure with the Cambridge Crystallographic Data Centre. The coordinates can be obtained, on request, from the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK.
(21) (a) van Dorp, D. A.; Word, J. P. Experientia. 1981, 37, 917. (b) Maurer, B.; Tohmmen, W. ibid. 1979, 62, 1096. (c) Maurer, B.; Grieder, A.; Tohmmen, W. Helv. Chim. Acta 1979, 62, 44.
(22) (a) Fujioka, H.; Kitagawa, H.; Kondo, M.; Kita, Y. Heterocycles 1994, 37, 743. (b) Fujioka, H.; Kitagawa, H.; Kondo, M.; Matsunaga, N.; Kitagaki, S.; Kita, Y. Heterocycles 1993, 35, 665.
(23) For examples, see: (a) Fadel. A.; Arzel, P. Tetrahedron: Asymmetry 1997, 8, 283. (b) Schoffers, E.; Golebiowski, A.; Johnson, C. R. Tetrahedron 1996, 52, 3769. (c) Theil, F. Chem. Rev. 1995, 95, 2203. (d) Banfi, L.; Guanti, G. Synthesis 1993, 1029. (e) Santaniello, E.; Ferraboschi, P.; Grisenti, P.; Manzocchi, A. Chem. Rev. 1992, 92, 1071. (f) Bolando, W.; Frobl, C.; Lorenz, M. Synthesis 1991, 1049.
(24) For recent examples for 1,2-diols, see: (a) Maezaki, N.; Sakamoto, A.; Soejima, M.; Sakamoto, I.; Xia, L.; Tanaka, T.; Ohishi, H.; Sakaguchi, K.; Iwata, C. Tetrahedron: Asymmetry 1996, 7, 2787. (b) Maezaki, N.; Soejima, M.; Takeda, M.; Sakamoto, A.; Matsumori, Y.; Tanaka, T.; Iwata, C. Tetrahedron 1996, 52, 6527. (c) Maezaki, N.; Soejima, M.; Sakamoto, A.; Sakamoto, I.; Matsumori, Y.; Tanaka, T.; Ishida, T.; In, Y.; Iwata, C. Tetrahedron: Asymmetry 1996, 7, 29. (d) Vedejs, E.; Daugulis, O.; Diver, S. T. J. Org. Chem. 1996, 61, 430. (e) Maezaki, N.; Soejima, M.; Takeda, M.; Sakamoto, A.; Tanaka, T.; Iwata, C. J. Chem. Soc. Chem. Commun. 1994, 1345. (f) Suemune, H.; Watanabe, K.; Kato, K.; Sakai, K. Tetrahedron: Asymmetry 1993, 4, 1767. (g) Harada, T.; Wada, I.; Oku, A. J. Org. Chem. 1989, 54, 2599. (h) Mukaiyama, T.; Tomioka, I.; Shimizu, M. Chem. Lett. 1984, 49. For recent examples for 1,3-diols, see: (a) Davis, A. P. Angew. Chem. Int. Ed. Engl. 1997, 36, 591. (b) Prasad, K.; Underwood, R. L.; Repic, O. J. Org. Chem. 1996, 61, 384. (c) Harada, T.; Oku, A. Synlett 1994, 95. (d) Suzuki, T.; Uozumi, Y.; Shibasaki M. J. Chem. Soc. Chem. Commun. 1991, 1593. (e) Appelt, A.; Willis, A. C.; Wild, S. B. J. Chem. Soc. Chem. Commun. 1988, 934. (f) Mukaiyama, T.; Tanabe, Y.; Shimizu, M. Chem. Lett. 1984, 401. (g) Ichikawa, J.; Asami, M.; Mukaiyama, T. Chem. Lett. 1984, 949. (h) Nara, M.; Terashima, S.; Yamada, S. Tetrahedrn 1980, 36, 3161. For recent example for 1,4-diols, see: Ishihara, K.; Kubota, M.; Yamamoto, H. Synlett 1994, 611.
(25) Prepared in three steps: i) aymmetric Diels-Alder reaction of the commercially available N-crotonyl-(4S)-isopropyl-2-oxazolidinone and cyclopentadiene according to Evans's method (Evans, D. A.; Chapman, K. T.; Bisaha, J. J. Am. Chem. Soc. 1988, 110, 1238.); ii) LiAlH4 reduction in THF at room temperature (97%); iii) Swern oxidation (73%).
(26) There are some reports showing that acetalization of an aldehyde and a cis-diol tends to form a cis-acetal as a major product under kineticcontrol. For review, see: Clode, D. M. Chem. Rev. 1979, 79, 491.
(27) Because it is known that cis-bicyclo[3.3.0]octane is more stable than the corresponding trans-isomer, two cis-intermediates B and B' are shown.
(28) Naemura, K.; Takeuchi, S.; Hirose, K.; Tobe, Y.; Kaneda, T.; Sakata, Y. J. Chem. Soc., Perkin Trans. 1 1995, 213.
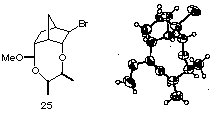
(29) X-ray analysis was performed on the mixed acetal 25 obtained by the same intramolecular haloetherification of the ene acetal derived from meso-2,3-butanediol 17e and (+)-5-norbornen-2-carboxyaldehyde.
(30) The ee value of silyl ether 24a was determined by 1H NMR analysis as its acetate because 24a was found to decompose under the HPLC analysis conditions. Ee values of 24b and 24c were determined by HPLC analysis (Chiralpak AD; hexane/iPrOH=93/7).
(31) Ene acetals 17 reported in this paper were obtained as a single isomer and their stereochemistry were determined to be cis by NOE experiment.
(32) Typical experimental procedure for dehaloetherification: To a stirred solution of 20i-l (0.1 mmol) in CH3CON(CH3)2 (1 ml) were added ZnCl2 (6 equiv.) and Zn (20 equiv.) under N2. The mixture was stirred at around 80ÁC. After completion of the reaction (TLC), the mixture was diluted with ether and filtered through a celite pad. The filtrate was evaporated in vacuo and purified by silica gel column chromatography (hexane/EtOAc) to afford 22i-l.