| [Related articles/posters: 036 109 012 ] |
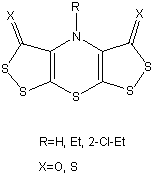
Computational Procedure.
An initial estimate of the geometry was obtained by molecular mechanics minimisation using the MM2 force field in a CAChe system. This approximate geometry was then re-optimised at the PM32 level using the MOPAC V6.0 program3 implemented on CAChe. An optimised PM3 sequential search over N6-C15-C18-Cl21 and C5-N6-C15-C18 angles was then carried out. The local minima found were then subjected to density functional theory ab initio calculation using the GAUSSIAN94 program4 system.
Results
A PM3 optimised search of the conformational space over N6-C15-C18-Cl21 and C5-N6-C15-C18 dihedrals produced three local minima: structure 1 (DH=24.22 Kcal/mol, N6-C15-C18-Cl21=180.0, C5-N6-C15-C18=-69.4), structure 2 (DH=24.51 Kcal/mol, N6-C15-C18-Cl21=81.8, C5-N6-C15-C18=-78.4) and structure 3 (DH=25.12 Kcal/mol, N6-C15-C18-Cl21=180.0, C5-N6-C15-C18=113.4)
The PM3 calculations predict the most stable conformation to be 1 whereas the crystal structure geometry most resembles 2. However, the difference in energy is less than 0.3 Kcal/mol which is within the error margin due to the calculation method.
The geometries of structures 1, 2 and 3 were then optimised using ab initio density functional theory with Becke's three parameter hybrid method B3LYP5. In this case, the results are in agreement with those from the crystallographic analysis. Conformation 2 (E[B3LYP/6-31G(d)]=-2963.5696423 Hartrees/particle, N6-C15-C18-Cl21=67.75, C5-N6-C15-C18=-94.13) is 1.51 Kcal/mol lower in energy than 1 (E[B3LYP/6-31G(d)]=-2963.567241 Hartrees/particle) and 1.79 Kcal/mol less energetic than 3 (E[B3LYP/6-31G(d)]=-2963.5667946 Hartrees/particle). Thus, there seems to be an attractive interaction between the chlorine atom and the ring that contributes to the crystal structure conformation and therefore, this is not only a consequence of the crystal packing.
The figure below shows both crystal structure6 and theoretical geometry and the superimposed result. Table 1 shows geometrical data for both of them.
Interestingly, when the substituent on the nitrogen atom is hydrogen, the crystal structure 6 of the molecule is planar, the only out of plane component being a minor displacement of the NH atom. At RHF, DFT and MP2 ab initio levels, a non-planar central ring is predicted. We were intrigued as to why an apparently 8p and hence formally anti-aromatic heterocycle, should nevertheless apparently prefer a planar geometry. To assess this aromaticity, the calculated7 nucleus independent chemical shift8 (NICS) on the DFT geometry was evaluated, and found to correspond to a non-aromatic (-0.59) system. The value found when the crystal structure geometry is used for the calculation is more antiaromatic (+4.64), but this value is relatively small compared to e.g. the value of cyclobutadiene (ca +20). This relatively small anti-aromaticity is probably the result of extensive push-pull cross conjugation in this ring system.
Regarding the discrepancy between the calculated and experimental geometries for the NH system, we strongly suspect that the planarity is induced by the p-p stacking exhibited by this system, which induces sufficient stabilisation to overcome the low energy barrier for inversion of the non-planar calculated form.
We thank Professor C. W. Rees and Dr T. Torroba for many valuable dicsussions, and Professor D. J. Williams and Dr A. J. P. White for details of the crystal structures.
1. C.F. Marcos, C. Polo, O.A. Rakitin, C.W. Rees and T. Torroba, Angew. Chem. Int. Engl., 1997, 36, 281. C.W. Rees, A. J. P. White, D. J. Wiliams, O. A. Rakitin, C. F. Marcos, C. Polo and T. Torroba, J. Org. Chem., 1998, 63, 2189.
2. J.J.P.Stewart, J. Comp. Chem., 1989, 10, 209 and J.J.P.Stewart, J. Comp. Chem., 1989, 10, 221.
3. J.J.P.Stewart, MOPAC, Quantum Chemistry Program Exchange, University of Indiana, Bloomington, USA, Program 455.
4. Gaussian program systems available from Gaussian Inc., Carnegie Office Park, Building 6, Pittsburgh, PA 15106 USA.
5. A.D. Becke, J.Chem.Phys., 1993, 98, 1293.
6. C.W. Rees, T. Torroba, D. J. Williams, personal communication.
7. GIAO-HF/6-31G(d)//B3LYP/6-31G(d).
8. P.v.R. Scheleyer,C. Maerker, A. Dransfeld, H. Jiao and N.J.R.vE. Hommes, J.Am.Chem.Soc., 1996, 118, 6317.