| [Molecules: None] [Related articles/posters: 052 007 018 044 056 ] |
The synthetic reaction using functionalized allylstannanes is widely appreciated as one of the most useful methods for the stereocontrolled C-C bond formation.1 Several methods have been reported for the diastereo- and enantioselective synthesis of 1,2-diol derivatives via the intermolecular reaction of g-alkoxyallylstannane with aldehydes.2 We now report the stereoselective synthesis of functionalized heterocycles via the intramolecular reaction of allylstannane with aldehyde and imine (eq 1).
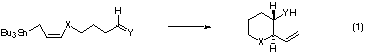
Total Synthesis of Hemibrevetoxin B
In recent years there has been an explosion of interest in biologically active natural products of marine origine.3 Due to their structural novelty and toxicity, polycyclic ethers are particularly attractive target for synthetic chemists.4 We have already reported a new strategy for the construction of cyclic ether via the intramolecular reaction of g-alkoxyallylstannane with aldehyde.5 The usufulness of this methodology is demonstrated by the total synthesis of marine natural product, hemibrevetoxin B (1).6,7
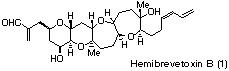
Hemibrevetoxin B (1), isolated from cultured cells of the red tide organism Gymnodinium breve by Y. Shimizu in 1989,8 has a 6,6,7,7-tetracyclic ether skeleton and contains 10 stereocenters. The preparation of 6,6-ring system was carried out primarily based on the modified Nicolaou's method (Scheme I). The mannose-derived stating material 2 was converted into 5 by benzylation followed by removal of the acetonide protection and selective elaboration of the liberated diol using Bu2SnO/BnBr and TESCl/imidazole. Ozonolysis of the double bond followed by treatment of the resulting aldehyde by a Wittig reagent afforded 6 in 91% yield. Reduction with diisobutyl-aluminium hydride gave allylic alcohol 7 in 87% yield, which was converted into the epoxide 8 upon treatment with the Sharpless epoxidation reagent. Oxidation of the primary alcohol of 8 with SO3*py-DMSO-Et3N followed by Wittig reaction afforded 9 in 82% overall yield. Removal of the TES protecting group by using tetrabutylammonium fluoride afforded 10 in quantitative yield. Ring opening and cyclization with camphorsulphonic acid gave 11 in 79% yield, which was converted to the acetate 12 in 95% yield.
Scheme Ia
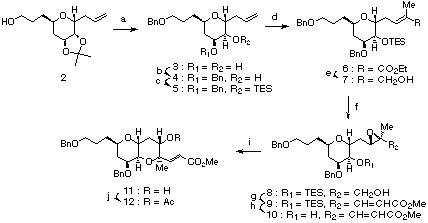
a(a) (i) BnBr, KH, THF, rt, 91%; (ii) HCl, MeOH, rt, 100%; (b) (i) Bu2SnO, MeOH, reflux; (ii) BnBr, CsF, DMF, rt, 80%; (c) TESCl, imidazole, DMF, rt, 91%; (d) (i) O3, CH2Cl2, -78 deg.C, then Ph3P, rt; (ii) Ph3P=C(Me)CO2Et, benzene, reflux, 91%; (e) DIBAL-H, CH2Cl2, -78 deg.C, 87%; (f) (+)-DET, Ti(OiPr)4, t-BuOOH, MS4A, CH2Cl2, -20 deg.C; (g) (i) SO3*py, DMSO, Et3N, CH2Cl2, rt; (ii) Ph3P=CHCO2Me, benzene, reflux (82% from three steps); (h) TBAF, THF, rt, 100%; (i) CSA, CH2Cl2, rt, 79%; (j) Ac2O, pyridine, DMAP, CH2Cl2, rt, 95%
Scheme IIa
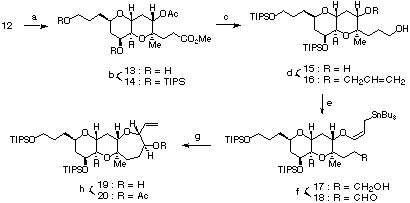
a(a) H2, Pd(OH)2-C, MeOH, rt, 82%; (b) TIPSOTf, 2,6-lutidine, DMF, rt to 70 deg.C, 97%; (c) LiAlH4, ether, 0 deg.C, 100%; (d) (i) TESCl, Et3N, CH2Cl2, -15 deg.C; (ii) allyl bromide, KH, THF, rt; (iii) Amberlyst-15, EtOH, rt, 83%; (e) sec-BuLi, TMEDA, THF, -78 deg.C, then Bu3SnCl, -78 deg.C to rt, 69%; (f) SO3*py, DMSO, Et3N, CH2Cl2, rt, 90%; (g) BF3*OEt2, CH2Cl2, -78 deg.C, 94%; (h) Ac2O, pyridine, DMAP, CH2Cl2, rt, 100%.
The stereocontrolled synthesis of the 6,6,7-ring system is shown in Scheme II. Debenzylation and hydrogenation of the double bond of 12 was achieved by using H2/Pd(OH)2-C to give 13 in 82% yield. The free OH groups were protected with TIPSOTf/2,6-lutidine to give 14 in 97% yield. Reduction of 14 with LiAlH4 afforded 15 in quantitative yield. The method for seven-membered ring formation based on allylic tin-aldehyde condensation was then used. Selective protection of the primary alcohol with TESCl/Et3N followed by allylation of the secondary alcohol and selective cleavage of the TES ether gave 16 in 83% yield. Formation of the corresponding allylic anion followed by trapping with Bu3SnCl afforded 17 in 69% yield. Oxidation with SO3*py/DMSO/Et3N produced 18 in 90% yield. Cyclization of 18 with BF3*OEt2 proceeded smoothly and stereoselectively to give 19 in 94% yield, which was converted to 20 by acetylation. Only one diastereoisomer was detected in the cyclization step.
Scheme IIIa
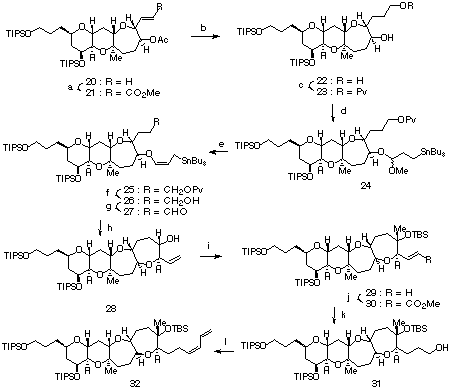
a(a) (i) O3, CH2Cl2, -78 deg.C, then Ph3P, -78 deg.C to rt; (ii) Ph3P=CHCO2Me, CH2Cl2, 0 deg.C to rt, 99%; (b) (i) H2, 10% Pd-C, AcOEt, rt; (ii) LiAlH4, ether, 0 deg.C, 98%; (c) PvCl, pyridine, CH2Cl2, 0 deg.C, 95%; (d) g-methoxyallylstannane, CSA, CH2Cl2, rt, 93%; (e) TMSI, HMDS, CH2Cl2, -15 deg.C, 85%; (h) BF3*OEt2, CH2Cl2, -78 deg.C, 98%; (i) (i) Swern oxidation; (ii) MeMgBr, ether, -78 deg.C to rt; (iii) TBSOTf, 2,6-lutidine, CH2Cl2, rt, 89% (ca. 1:1 mixture of isomers); (j) (i) O3, CH2Cl2, -78 deg.C, then Ph3P, -78 deg.C to rt; (ii) Ph3P=CHCO2Me, benzen, reflux, 78%; (k) (i) H2, 10% Pd-C, AcOEt, rt; (ii) LiAlH4, ether, 0 deg.C, 92%; (l) (i) Dess-Martin periodinane, CH2Cl2, rt; (ii) PhSe(CH2)3Ph3P+I-, n-BuLi, HMPA, -78 deg.C to rt; (iii) H2O2, NaHCO3, THF, rt, 52%.
The stereoselective construction of the 6,6,7,7-ring system is shown in Scheme III. Ozonolysis of 20 followed by chain elongation gave 21 in 99% yield. Reduction with H2/Pd-C and LiAlH4 afforded 22 in 98% yield. Selective protection of the primary alcohol with PvCl/pyridine gave 23 in 95% yield. The reaction of 23 with g-methoxyallylstannane in the presence of a catalytic amount of CSA afforded mixed acetal 24 in 93% yield. The elimination of methanol from 24 proceeded smoothly by the treatment with TMSI/HMDS to give 25 in 85% yield.9 Due to the steric bulkiness and the high functionalization of the substrate, only poor result was obtained by the usual allylic anion formation followed by trapping with n-Bu3SnCl.6 Deprotection followed by oxidation gave 27. The BF3*OEt2 mediated cyclization of 27 afforded 28 as a sole product in 98% yield. Oxidation of 28, Grignard reaction with MeMgBr, and TBS protection gave a 1:1 mixture of epimeric isomers in 89% yield, from which the desired isomer 29 was isolated by chromatography.10 Ozonolysis of 29 followed by Wittig reaction gave 30 in 78% yield. Reduction with H2/Pd-C and LiAlH4 afforded 31 in 92% yield. Dess-Martin oxidation of 31 followed by treatment with the ylid derived from PhSe(CH2)3Ph3P+I- and n-BuLi, and oxidation-syn-elimination using H2O2 and NaHCO3 afforded diene 32 in 52% yield.7a
The final stage of our synthetic study is shown in Scheme IV. Selective desilylation of 32 using TBAF gave 33 in 73% yield. Dess-Martin oxidation followed by treatment with Eschenmoser's salt afforded 34 in 55% yield.11 Finally, the silyl protecting groups were removed by using SiF4 to give hemibrevetoxin B (1) in 68% yield. The 1H and 13C-NMR spectra of synthetic hemibrevetoxin B (1) were identical with those of the natural product.
Scheme IVa
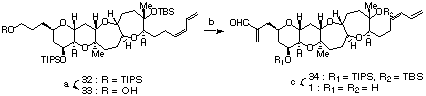
a(a) TBAF, THF, rt, 73%; (b) (i) Dess-Martin periodinane, CH2Cl2, rt; (ii) Me2(CH2)N+I-, Et3N, CH2Cl2, rt, 55%; (c) SiF4, CH2Cl2-CH3CN (1:1), rt, 68%.
Intramolecular Reaction with Imine Derivatives
To extend our methodology, we next examined the intramolecular reaction with imine,12 because the allylation of imine has been well studied as well as that of aldehyde,1 and such transformation would be an efficient method for the synthesis of cyclic amine derivatives.
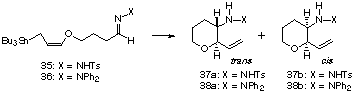
After several fruitless attempts, we found hydrazones are suitable as a carbon-nitrogen double bond functional group for the intramolecular reaction. The cyclization substrates 35 and 36, g-alkoxyallylstannanes having a hydrazone group at the terminus of the carbon chain, were easily prepared by the reaction of the corresponding aldehyde with hydrazines, and could be purified by silica gel column chromatography without any trouble. The results of the cyclization of 35 and 36 are summarized in Table 1. In all cases of the Lewis acid mediated reactions, trans isomers 37a and 38a were obtained as a sole product in high to good yields (entries 2, 4-7, and 11-15). None of cis isomers 37b and 38b could be detected by 1H NMR analysis of the crude product. Although the use of TiCl4 caused decomposition of the substrate (entry 1), TiCl2(OiPr)2 promoted the cyclization of tosylhydrazone 35 at -78 deg.C to give 37a in 94% yield (entry 2). No reaction took place with Ti(OiPr)4 even at room temperature, presumably due to its low Lewis acidity (entry 3). The use of Lewis acids, such as ZrCl4, AlCl3, ZnCl2, and BF3*OEt2, gave lower yields of the product (entries 4-7). Although the reaction proceeded quantitatively in the presence of protic acids such as CF3SO3H and CF3CO2H, the trans selectivity was decreased to the ratio of ca. 7:3 (entries 8 and 9). These results suggested that the thermal cyclization of 35 would proceed via cyclic transition state to give cis isomer 37b with high stereoselectivity. However, unfortunately, only decomposition of 35 took place when it was refluxed in toluene (entry 10). Diphenylhydrazone 36 also cyclized in the presence of TiCl2(OiPr)2 to give trans isomer 38a quantitatively (entry 11). Interestingly, the use of a catalytic amount of Yb(OTf)3 (0.5 eq) promoted the cyclization to give 38a with high stereoselectivity in high yield (entry 12).14 The use of smaller amount of the lanthanide catalysts decreased the conversion yield (entries 13-15).
Table 1. Cyclization of 35 and 36a
| entry | substrate | reagent | equiv | temp (deg.C) | time (min) | ratio (trans:cis) | yield(%)b |
| 1 | 35 | TiCl4 | 2.0 | -78 | 5 | decomposition | |
| 2 | TiCl2(OiPr)2 | 2.0 | -78 | 5 | >95:5 | 94 | |
| 3 | Ti(OiPr)4 | 2.0 | rt | 30 | no reaction | ||
| 4 | ZrCl4 | 2.0 | -78 | 180 | >95:5 | 81 | |
| 5 | AlCl3 | 2.0 | -78 | 120 | >95:5 | 68 | |
| 6 | ZnCl2 | 2.0 | -6 | 180 | >95:5 | 71 | |
| 7 | BF3*OEt2 | 2.0 | -78 | 5 | >95:5 | 61 | |
| 8 | CF3SO3H | 2.0 | -78 | 30 | 71:29 | 98 | |
| 9 | CF3CO2H | 2.0 | -78 | 90 | 70:30 | 97 | |
| 10 | -c | - | 120 | 60 | decomposition | ||
| 11 | 36 | TiCl2(OiPr)2 | 2.0 | -78 5 | >95:5 | 97 | |
| 12 | Yb(OTf)3 | 0.5 | rt | 1080 | >95:5 | 97 | |
| 13 | Yb(OTf)3 | 0.2 | rt | 2880 | >95:5 | 81(16) | |
| 14 | Yb(OTf)3 | 0.2 | 40 | 2400 | >95:5 | 75(24) | |
| 15 | La(OTf)3 | 0.2 | rt | 2880 | >95:5 | 71(29) | |
Stereoselective Synthesis of Hydroxylated Pipridine and Pyrrolidine Derivatives
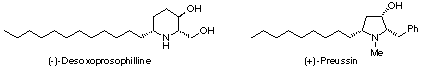
The structural framework of hydroxylated nitrogen heterocycle is widely found in naturally occurring piperidine/pyrrolidine alkaloids such as (-)-desoxoprosophilline15 and (+)-preussin.16 We examined the stereoselective synthesis of b-hydroxypiperidine and pyrrolidine derivatives via the intramolecular reaction of g-aminoallylstannane with aldehyde.17 To the best of our knowledge, this is the first example of a successful use of g-aminoallylstannane in organic synthesis.18
The results of the cyclization of 39 are summaried in Table 2. The use of TiCl4 and/or BF3*OEt2 gave trans-b-hydroxypiperidine 40a, predominantly (entries 1 and 10). Interestingly, although the reason is not clear, the reaction mediated by titanium chloride (entries 2-4), tin chloride (entries 7 and 8), and ZnI2 (entry 14) afforded cis isomer 40b as a major product. Similar cis selectivity was observed in the protic acid promoted cyclization (entries 15 and 16) which would proceed via a cyclic transition state as proposed previously. The cyclization of 39 did not proceed in the presence of weak Lewis acids such as Ti(OiPr)4 (entry 5) and Bu2SnCl2 (entry 9). As expected, the thermal reaction afforded 40b with very high stereoselectivity (entry 17).
Table 2. Cyclization of 39a
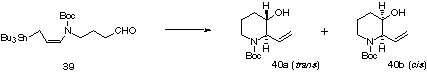
| entry | reagent | (equiv) | temp (deg.C) | time (h) | ratio (40a:40b)b | yield (%)c |
| 1 | TiCl4 | (1.5) | -78 | 0.7 | 69:31 | 63 |
| 2 | TiCl3(OiPr) | (1.5) | -78 | 0.5 | 38:62 | 46 |
| 3 | TiCl2(OiPr)2 | (1.5) | -78 | 1.5 | 29:71 | 89 |
| 4 | TiCl(OiPr)3 | (1.5) | rt | 8.0 | 31:69 | 56 |
| 5 | Ti(OiPr)4 | (3) | rt | 24 | - | 0d |
| 6 | ZrCl4 | (1.5) | -78 | 2.5 | 53:47 | 65 |
| 7 | SnCl4 | (1.5) | -78 | 1.0 | 34:66 | 41 |
| 8 | BuSnCl3 | (1.5) | -78 | 0.5 | 28:72 | 20 |
| 9 | Bu2SnCl2 | (1.0) | rt | 15 | - | 0d |
| 10 | BF3*OEt2 | (2.0) | -78 | 0.5 | 70:30 | 60 |
| 11 | AlCl3*OEt2 | (1.2) | -78 | 0.5 | 64:36 | 45 |
| 12 | AlBr3*OEt2 | (1.5) | -78 | 0.7 | 50:50 | 69 |
| 13 | MgBr2*OEt2 | (1.5) | 0 | 2.0 | 61:39 | 92 |
| 14 | ZnI2 | (1.5) | rt | 7.0 | 36:64 | 45 |
| 15 | HCl | (2.0) | -78 | 1.0 | 27:73 | 48 |
| 16 | CF3SO3H | (2.0) | -78 | 3.0 | 43:57 | 21 |
| 17 | -e | 120 | 36 | 2:98 | <67 | |
We next examined the synthesis of pyrrolidine derivative using 41. All reactions were carried out immediately after preparation of the substrate 41 owing to its low stability. The results are summarized in Table 3. Although the Lewis and protic acid mediated reactions gave unsatisfactory results, very high stereoselectivity was observed in the thermal cyclization. Trans isomer 42a was not detected. The stereoselective formation of the cis isomer 42b is highly promising as a methodology for the synthesis of nitrogen heterocycles, since (+)-preussin possesses cis-stereochemistry between a- and b-substituents.
Table 2. Cyclization of 41a
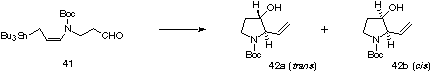
| entry | reagent (equiv) | temp (deg.C) | time (h) | ratio (42a:42b)b | yield (%)c |
| 1 | TiCl4 (1.2) | -78 | 0.5 | 10:90 | 48 |
| 2 | BF3*OEt2 (1.2) | -78 | 0.5 | 64:36 | 70 |
| 3 | CF3CO2H (2.0) | -78 | 0.5 | 49:51 | 70 |
| 4 | -d | 120 | 1.0 | 2:98 | < 67 |
2. (a) Yamamoto, Y.; Saito, Y.; Maruyama, K. Tetrahedron Lett. 1982, 23, 4959-4962. (b) Keck, G. E.; Abbott, D. E.; Wiley, M. R. Tetrahedron Lett. 1987, 28, 139-142. (c) Koreeda, M.; Tanaka, Y. Tetrahedron Lett. 1987, 28, 143-146. (d) Marshall, J. A.; Gung, W. Y. Tetrahedron Lett. 1989, 30, 2183-2186. (e) Marshall, J. A.; Luke, G. P. J. Org. Chem. 1991, 56, 483-485. (f) Yamamoto, Y.; Kobayashi, K.; Okano, H.; Kadota, I. J. Org. Chem. 1992, 57, 7003-7005. (g) Roush, W. R.; VanNieuwenhze, M. S. J. Am. Chem. Soc. 1994, 116, 8536-8543. (h) Kadota, I.; Kobayashi, K.; Okano, H.; Asao, N. Bull. Soc. Chim. Fr. 1995, 132, 615-623.
3. For recent reviews, see: (a) Shimizu, Y. Chem. Rev. 1993, 93, 1685-1698. (b) Yasumoto, T.; Murata, M. Chem. Rev. 1993, 93, 1897-1909.
4. For recent review, see: Alvarez, E.; Candenas, M.-L.; Pérez, R.; Ravelo, J. L.; Martín, J. D. Chem. Rev. 1995, 95, 1953-1980, and references cited therein.
5. (a) Yamamoto, Y.; Yamada, J.; Kadota, I. Tetrahedron Lett. 1991, 32, 7069-7072. (b) Gevorgyan, V.; Kadota, I.; Yamamoto, Y. Tetrahedron Lett. 1993, 34, 1313-1316. (c) Yamamoto, Y.; Kadota, I. Main Group Met. Chem. 1994, 17, 269-289.
6. Kadota, I.; Park, J.-Y.; Koumura, N.; Pollaud, G.; Matsukawa, Y.; Yamamoto, Y. Tetrahedron Lett. 1995, 36, 5777-5780.
7. First total synthesis: (a) Nicolaou, K. C.; Reddy, K. R.; Skokotas, G.; Sato, F.; Xiao, X.-Y.; Hwang, C.-K. J. Am. Chem. Soc. 1993, 115, 3558. (b) Nicolaou, K. C.; Reddy, K. R.; Skokotas, G.; Sato, F.; Xiao, X.-Y.; Hwang, C.-K. J. Am. Chem. Soc. 1992, 114, 7935. Synthetic studies: (c) Feng, F.; Murai, A. Chem. Lett. 1992, 1587-1590. (d) Feng, F.; Murai, A. Chem. Lett. 1995, 23-24. (e) Kadota, I.; Matsukawa, Y.; Yamamoto, Y. J. Chem. Soc., Chem. Commun. 1993, 1638-1641. (f) Yamamoto, Y.; Kadota, I. Bull. Soc. Chim. Belg. 1994, 103, 619-629. (g) Feng, F.; Murai, A. Synlett 1995, 863-865. (h) Nakata, T.; Nomura, S.; Matsukura, H.; Morimoto, M. Tetrahedron Lett. 1996, 37, 217-220.
8. Prasad, A. V. K.; Shimizu, Y. J. Am. Chem. Soc. 1989, 111, 6476-6477.
9. Kadota, I.; Sakaihara, T.; Yamamoto, Y. Tetrahedron Lett. 1996, 37, 3195-3198.
10. We examinded the cyclization of the allylic tin-methyl ketone (derived from 27), since previously this type of cyclization proceeded smoothly.7e,f However, such attempts resulted in a failure in this case.
11. Takano, S.; Inomata, K.; Samizu, K.; Tomita, S.; Yanase, M.; Suzuki, M.; Iwabuchi, Y.; Sugihara, T.; Ogasawara, K. Chem. Lett. 1989, 1283-1284.
12. Kadota, I.; Park, J.-Y.; Yamamoto, Y. J. Chem. Soc., Chem. Commun., 1996, 841-842.
13. Catalytic intermolecular condensation of allylstannane and imine by using lanthanide triflates has been reported; see: Bellucci, C.; Cozzi, P. G.; Umani-Ronchi, A. Tetrahedron Lett. 1995, 36, 7289-7292.
14. Khuong-Huu, Q.; Ratle, G.; Monseur, X.; Goutarel, R. Bull. Soc. Chim. Belg. 1972, 81, 425.
15. (a) Schwartz, R. E.; Liesch, J.; Hensens, O.; Zitano, L.; Honeycutt, S.; Garrity, G.; Fromtling, R. A.; Onishi, J.; Monaghan, R. J. Antibiot. 1988, 41, 1774. (b) Johnson, J. H.; Phillipson, D. W.; Kahle, A. D. J. Antibiot. 1989, 42, 1184.
16. Kadota, I.; Kawada, M.; Saya, S.; Yamamoto, Y. Tetrahedron Lett., 37, 2109-2112(1996).
17. In general, g-aminoallylstannanes exhibit lower reactivity toward aldehydes than g-alkoxyallylstannanes.