| [Molecules: 11] [Related articles/posters: 038 061 054 114 043 ] |
This turned out not to be the case.
Reactions
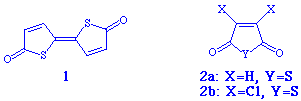
When the thioanhydride 2a was allowed to react with trimethyl or triethyl phosphite in methylene chloride at or below room temperature (Scheme 1, reaction A), the spiro compound 3 could be isolated in 10% yield. NMR analysis of the crude product indicated, however, an almost quantitative conversion to 3. Extensive decomposition during work-up accounts for the low isolated yield.
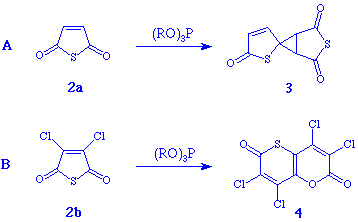
The structure of 3 was determined by NMR, MS and FTIR and verified by X-ray
crystallography. See experimental details.
The compound crystallises with two independent molecules in the asymmetric unit,
or four molecules in
the triclinic unit cell.
When the dichloro-substituted thioanhydride 2b was similarly treated
with trimethyl or triethyl phosphite in methylene chloride (Scheme 1, reaction B),
the reaction took a totally different course, producing the compound 4 in 20%
isolated yield. The structure was determined by X-ray crystallography and verified
spectroscopically (NMR, MS and FTIR). See experimental details.
The compound forms a disordered crystal, with ring S or O statistically occupying almost the same positions.
Here is an attempted resolution of the overlapping structures.
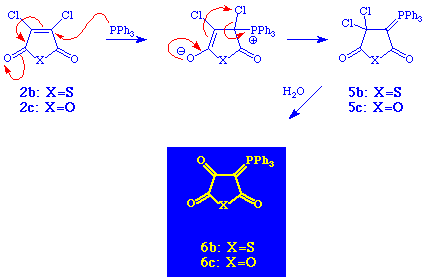
When triphenyl phosphine was used instead of trialkyl phosphites, the outcome of the reaction was quite different (Scheme 2). The chloro-substituted compounds 2b and 2c now gave the ylids 5b and 5c, respectively, which after hydrolysis afforded 6b and 6c both in total yields of 71% with melting points of 190-193 and 234-235 oC, respectively. See experimental details.
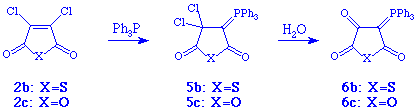
The products were characterised spectroscopically (NMR and
MS) and
the structure of 6c was determined by X-ray crystallography.
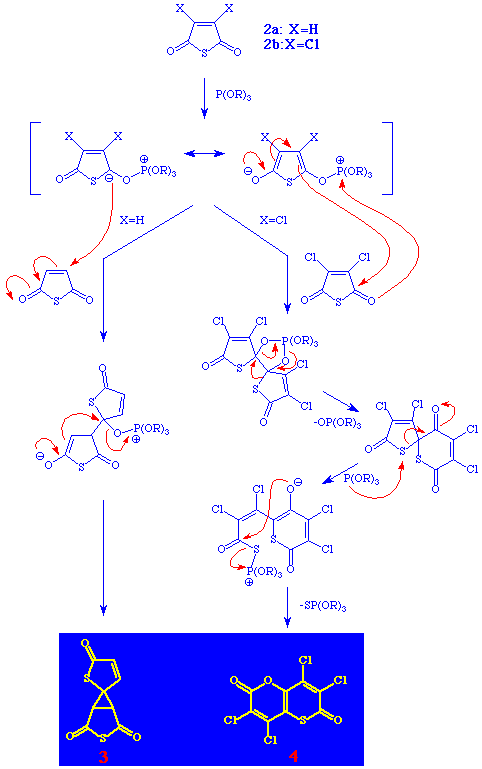
There is ample evidence in the literature that trivalent phosphorus compounds react with p-quinones or cyclic unsaturated anhydrides, producing oxyphosphoranes via radical intermediates.5
In Scheme 3 this is depicted (in parentheses) as formation of resonance stabilised zwitterionic oxyphosphoranes.
When the thioanhydride is unsubstituted (X=H), the next step is a nucleophilic attack of the carbanionic centre in the 2-position of the oxyphosphorane at the 3-position of the thioanhydride producing a zwitterionic Michael adduct. Elimination of trialkyl phosphate affords the spiro compound 3.
When, on the other hand, the thioanhydride is substituted (X=Cl), there seem to be steric (and possibly electronic) effects precluding the formation of the spiro compound. Instead we suggest a primary attack on one of the carbonyl carbons with simultaneous formation of a bond between the carbonyl oxygen and the phosphor atom. An analogue to this cyclic five-coordinated oxyphosphorane thus formed has previously been suggested by Markgraf in a similar reaction from thiophthalic anhydride.2 The next steps involve elimination of trialkyl phosphate with ring expansion of one of the sulfur-containing rings. An attack of trialkyl phosphite on a sulfur atom produces a zwitterion from which compound 4 is formed with simultaneous elimination of trialkyl thiophosphate. The suggested mechanism is supported by the gas chromatographic detection of trialkyl phosphate and trialkyl thiophosphate in the reaction mixture.
As shown in Scheme 2 the reaction between triphenyl phosphine and the chloro-substituted anhydrides 2b and 2c took a quite different course. The initial step in this reaction appears to be a Michael type addition to produce a zwitterionic phosphorane which then undergoes a chloronium rearrangement with formation of phosphorus ylides 5b and 5c as depicted in Scheme 4. Subsequent hydrolysis gives 6b and 6c.
We suggest the following reason, but welcome other explanations.
The reactions reported in Scheme 1 and explained in Scheme 3 involve radicals in the initial steps. These radicals are initially produced by one-electron transfer from phosphorus to oxygen. The ease of such transfer should increase with the stability of the formed phosphinium radical cation. We suggest that trialkoxy phosphinium radical cations are more stable than the corresponding triphenyl phosphinium radical cation due to better resonance stabilisation in the former case. In fact there is a suggestion in the literature that the triphenyl phosphinium radical cation is not formed in a similar reaction with chloranil.5
We are still very much interested in the synthesis of 1. Suggestions will be received with gratitude.