Question:
Why is it possible to easily transmetalate primary
and cyclic secondary a-aminoorganostannanes, but not secondary acyclic
ones?
Hypothesis No. 1:
Conformational considerations in
the acyclic compounds raise the energy of activation, making the transmetalation
impossibly slow.
Background
It is known that cyclic, secondary a-aminoorganostannanes
will undergo transmetalation if there is a chelating group available nearby
(eqns. 10 and 11). [
1-5] This suggests that perhaps delivery of the alkyllithium by
prior coordination to the chelating atom (a complex-induced proximity effect)
[6] assists the transmetalation.
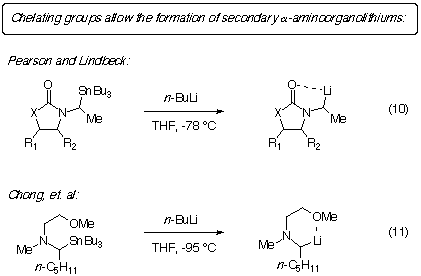
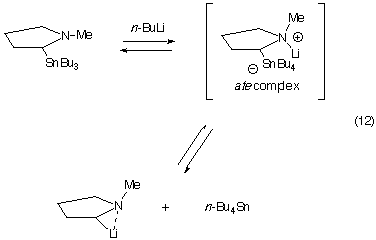
The mechanism of tin-lithium exchange
involves addition of a butyl group to the tin, making an ate-complex, which
then expels one of the alkyl groups on the tin as an organolithium. [7] This is an equilibrium process, where the most stable organolithium
is formed. [8] Thus, if one group
bears a heteroatom that can coordinate to lithium, this group might dissociate
from the ate-complex preferentially. The tin-lithium exchange occurs with
retention of configuration. [8,9]
Thus, if a heteroatom delivers the lithium to the carbon bearing the tin,
these two atoms must be in close proximity. In our work, in the absence
of a chelating group, the only heteroatom is the nitrogen. Thus, the lithium
may be coordinated to the nitrogen in the ate-complex, as shown in eqn.
12 for a 2-(tributylstannyl)pyrrolidine transmetalation. In support of this
notion is the fact that at least one 'unstabilized' a-aminoorganolithium
compound has the lithium bridged between the carbon and nitrogen in the
solid state. [10]
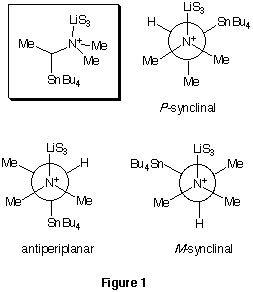
Note that the dihedral angle between the carbon-tin bond and
the nitrogen-lithium bond in this complex is <60° (i.e. synclinal)
in the above transmetalation. Could it be that such a synclinal arrangement
is not available to a secondary acyclic a-aminoorganostannane? The
Newman projections in Figure 1 illustrate the conformations of the ate complex
of a secondary a-aminoorganostannane (Bu4SnCHMeN+Me2LiS3), with a solvated
lithium (LiS3) coordinated to the nitrogen. If the antiperiplanar conformation
were considerably more stable than either of the synclinal conformations,
as would be expected if steric effects were dominant and LiS3 and SnBu4
were the largest groups, then the homofacial intramolecular delivery of
lithium to the carbanionic carbon would be impossible.
To test this possibility, we carried out ab initio calculations on
a model system, H4SnCHMeN+Me2Li(OH2)3, using the RHF/3-21G(d) basis set.
We examined the antiperiplanar and the P-synclinal conformations. The fully
minimized structures are shown in Figure 2. However, the antiperiplanar
conformation was found to be 19 kcal mol-1 (1 cal = 4.184 J) less stable than the P-synclinal
conformation, refuting the hypothesis.
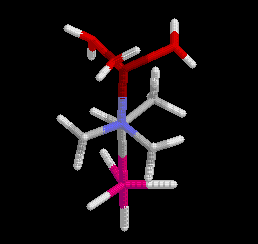
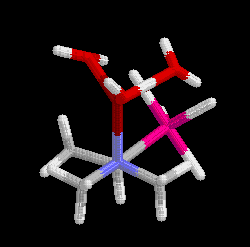
Figure 2 Antiperiplanar (left) and P-synclinal (right) conformations
Click on the structures to go to a rotatable picture.
Hypothesis No. 2
There is a difference in the thermodynamic
stabilities between cyclic and acyclic a-aminoorganolithiums that prevents
loss of the latter from an ate complex.
If the ate complex can be formed but the
transmetalation does not proceed, then the product must be less stable than
butyllithium. Although it is not obvious why there should be an
inherent difference in stability between cyclic and acyclic a-aminoorganolithiums,
we calculated the heats of formation of two isomeric lithium derivatives
of N-ethylpyrrolidine (Figure 3). At the RHF/3-21G level of theory
(GAUSSIAN94), N-ethyl-2-lithiopyrrolidine is 2.51 kcal mol-1 more
stable than N-(1-lithio)ethylpyrrolidine. Interestingly, both of
these compounds show significant bridging of the lithium across both carbon
and nitrogen, in accord with previous theoretical [11] and crystallographic [10]
results. The 3D structures are shown in Figure 4.
This work also allows us to rank the relative stability of various organolithiums.
Click here to see this ranking.
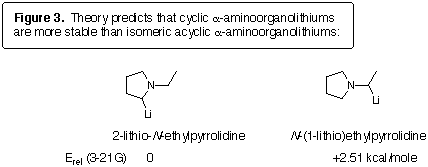
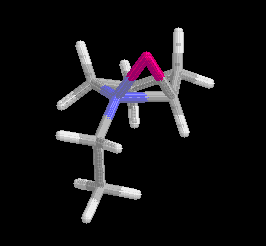

Figure 4 Calculated structures of 2-lithio-N-ethylpyrrolidine
(left) and N-(1-lithio)ethylpyrrolidine (right).
Click on the structures to go to a rotatable picture.
Where does this leave us?
The answer to the question posed
in the title appears to be that there is a sufficiently large difference
in the thermodynamic stability of the products that cyclic a-aminoorganolithiums
may be formed by tin-lithium exchange, but acyclic ones cannot. In other
words, butyllithium is more stable than the the acyclic
a-lithioorganolithium compound, but less stable than the cyclic
a-lithioorganolithium compound.
Why is this true? What is so special about having the organolithium in a
ring? It's a mystery. We would appreciate your comments.
Experiments to help answer this question are in progress and will be reported
in due course.
References
- 1. Pearson, W. H.; Lindbeck, A. C. J. Org
Chem. 1989, 54, 5651. Go
back to text.
- Pearson, W. H.; Lindbeck, A. C. J. Am. Chem. Soc. 1991,
113, 8546. Go back to text.
-
Pearson, W. H.; Lindbeck, A. C.; Kampf, J. W. J. Am. Chem. Soc.
1993, 115, 2622. Go back to text.
-
Chong, J. M.; Park, S. B. J. Org. Chem., 1992, 57,
2220. Go back to text.
-
Burchat, A. F.; Chong, J. M.; Park, S. B. Tetrahedron Lett. 1993,
34, 51. Go back to text.
-
Beak, P.; Meyers, A. I. Acc. Chem. Res. 1986, 19,
356. Go back to text.
-
Abraham, M. H. In Comprehensive Chemical Kinetics; ed. Bamford, C.
H., Tipper, C. F. H., Elsevier, Amsterdam, 1973; Vol. 12, pp. 1-256.
Go back to text.
-
Sawyer, J. S.; Kucerovy, A.; Macdonald, T. L.; McGarvey, G. J. J.
Am. Chem. Soc. 1988, 110, 842. Go
back to text.
-
Still, W. C.; Sreekumar, C. J. Am. Chem. Soc. 1980, 102,
1201. Go back to text.
-
Boche, G.; Marsch, M.; Harbach, J.; Harms, K.; Ledig, B.; Schubert,
F.; Lohrenz, J. C. W.; Ahlbrecht, H. Chem. Ber. 1993, 126,
1887. Go back to text. Go
back to later text.
-
Schleyer, P. v. R.; Clark, T.; Kos, A. J.; Spitznagel, G. W.; Rohde,
C.; Arad, D.; Houk, K. N.; Rondan, N. G. J. Am. Chem. Soc. 1984,
106, 6467. Go back to text.
Back to the introductory page.
1. Transmetalation of acyclic aminostannanes:
Primary organolithiums can be formed, but secondary organolithiums cannot
2. Transmetalation of cyclic aminostannanes:
Secondary organolithiums are formed
3. Hypotheses: Conformational effects (i.e., a kinetic problem) or thermodynamics
may be responsible. (THIS PAGE)
4. These studies allow a ranking of the relative
stabilities of organolithiums
5. How the aminostannanes were made